Amenorrhea pathophysiology: Difference between revisions
Akshun Kalia (talk | contribs) No edit summary |
m (Bot: Removing from Primary care) |
||
| (45 intermediate revisions by 3 users not shown) | |||
| Line 5: | Line 5: | ||
==Overview== | ==Overview== | ||
Amenorrhea is defined as absence of [[menstrual cycle]]. The | Amenorrhea is defined as absence of [[menstrual cycle]]. The causes of amenorrhea include [[hypothalamic]], [[pituitary]], [[thyroid]], [[adrenal]], [[ovarian]], [[uterine]], and [[vaginal]]. About 25 different [[genes]] are involved in the pathogenesis of amenorrhea including 3 different groups of [[Kallman syndrome|Kallmann syndrome]] related genes, [[hypothalamus]]-[[pituitary]]-[[gonadal]] (HPG) axis related genes, and [[obesity]] related [[genes]]. On [[gross pathology]], normal [[endometrium]] is the characteristic findings of amenorrhea. Patients of amenorrhea from [[Craniopharyngioma]] as have cystic mass filled with motor oil-like fluid on [[gross pathology]]. On [[microscopic]] [[histopathological]] analysis, [[craniopharyngioma]] presents as trabecular [[squamous epithelium]] surrounded by palisaded [[columnar epithelium]], small-to-medium sized cells with moderate amount of [[basophilic]] [[cytoplasm]], bland [[nuclei]], and [[Calcification|calcifications]]. On [[microscopic]] [[histopathological]] analysis, [[pituitary adenoma]] as a cause of amenorrhea presents as loss of [[fibrous]] [[stroma]] and nested cells of normal [[anterior pituitary]] (based on the type of [[adenoma]]). | ||
==Pathophysiology== | ==Pathophysiology== | ||
| Line 19: | Line 19: | ||
** During [[puberty]] the amplitude and frequency of [[GnRH]] pulses is significantly increased. | ** During [[puberty]] the amplitude and frequency of [[GnRH]] pulses is significantly increased. | ||
** [[GnRH]] secretion is regulated by certain [[neurotransmitters]] in [[brain]], such as [[dopamine]], endogenous [[opioids]], [[norepinephrine]], [[Gamma aminobutyric acid|gamma amino butyric acid (GABA)]], and [[Corticotropin releasing hormone|corticotropin releasing hormone (CRH)]]. Alteration in level and function of these [[neurotransmitters]] may lead to specific types of amenorrhea. For example [[stress]], [[exercise]], and [[malnutrition]] affects [[CRH]], [[β-endorphin]], and [[dopamine]], respectively.<ref name="pmid18574222">{{cite journal |vauthors=Golden NH, Carlson JL |title=The pathophysiology of amenorrhea in the adolescent |journal=Ann. N. Y. Acad. Sci. |volume=1135 |issue= |pages=163–78 |year=2008 |pmid=18574222 |doi=10.1196/annals.1429.014 |url=}}</ref> | ** [[GnRH]] secretion is regulated by certain [[neurotransmitters]] in [[brain]], such as [[dopamine]], endogenous [[opioids]], [[norepinephrine]], [[Gamma aminobutyric acid|gamma amino butyric acid (GABA)]], and [[Corticotropin releasing hormone|corticotropin releasing hormone (CRH)]]. Alteration in level and function of these [[neurotransmitters]] may lead to specific types of amenorrhea. For example [[stress]], [[exercise]], and [[malnutrition]] affects [[CRH]], [[β-endorphin]], and [[dopamine]], respectively.<ref name="pmid18574222">{{cite journal |vauthors=Golden NH, Carlson JL |title=The pathophysiology of amenorrhea in the adolescent |journal=Ann. N. Y. Acad. Sci. |volume=1135 |issue= |pages=163–78 |year=2008 |pmid=18574222 |doi=10.1196/annals.1429.014 |url=}}</ref> | ||
* | * After the onset of [[puberty]], the [[negative feedback]] on [[GnRH]] is removed. | ||
* Pulsatile secretion of [[GnRH]] induce [[LH]] and [[FSH]] production which finally lead to [[ovulation]]. | |||
* In the absence of [[Fertilization|fertilisation]], the ovarian [[follicle]] is turned into [[corpus luteum]]. [[Endometrium]] proliferates through [[estrogen]] release from [[corpus luteum]]. Withdrawal of [[progesterone]] from [[estrogen]]-mediated proliferated [[endometrium]] results in [[Menstrual cycle|menstrual]] [[bleeding]]. | |||
==== Hypothalamic-pituitary-ovarian (HPO) axis maturation ==== | ==== Hypothalamic-pituitary-ovarian (HPO) axis maturation ==== | ||
* After activation of the [[Hypothalamic-pituitary-gonadal axis|HPO axis]] during | * After activation of the [[Hypothalamic-pituitary-gonadal axis|HPO axis]] during second [[trimester]] of [[pregnancy]], the level of [[gonadotropins]] rises from mid to term [[pregnancy]]. After cessation of [[placental]] [[hormone]] [[feedback]], [[FSH]] and [[LH]] increases again slightly to mild secondary peak. | ||
* [[ | * Before [[puberty]], [[negative feedback]] from [[adrenal]] [[androgens]] keep the [[gonadotropins]] in low plasma level.<ref name="pmid9238251">{{cite journal |vauthors=Apter D |title=Development of the hypothalamic-pituitary-ovarian axis |journal=Ann. N. Y. Acad. Sci. |volume=816 |issue= |pages=9–21 |year=1997 |pmid=9238251 |doi= |url=}}</ref> | ||
* Right before [[puberty]], the sensitivity of [[hypothalamus]] to [[ | * Right before [[puberty]], the sensitivity of [[hypothalamus]] to [[negative feedback]] from [[adrenal]] [[androgens]] is decreased. This leads to increased production of GnRH from hypothalamus and make it possible for [[GnRH]] to be raised in magnitude and frequency to induce an increase in [[LH]] and [[FSH]].<ref name="pmid4852310">{{cite journal |vauthors=Boyar RM, Rosenfeld RS, Kapen S, Finkelstein JW, Roffwarg HP, Weitzman ED, Hellman L |title=Human puberty. Simultaneous augmented secretion of luteinizing hormone and testosterone during sleep |journal=J. Clin. Invest. |volume=54 |issue=3 |pages=609–18 |year=1974 |pmid=4852310 |pmc=301594 |doi=10.1172/JCI107798 |url=}}</ref> | ||
* The complete maturation of [[Hypothalamic-pituitary-gonadal axis|HPO axis]] takes around 5-7 years from [[menstruation]] | * The complete maturation of [[Hypothalamic-pituitary-gonadal axis|HPO axis]] takes around 5-7 years from the onset of [[menstruation]]. Generally, during the first two years of [[menstruation]], the cycles are mostly [[Anovulatory cycle|anovulatory]] due to immature HPO axis. | ||
===Pathogenesis=== | ===Pathogenesis=== | ||
*Amenorrhea is defined as absence of [[menstrual cycle]]. Primary amenorrhea is absence of [[ | *Amenorrhea is defined as the absence of [[menstrual cycle]].<ref name="pmid18574222" /> | ||
*The [[pathophysiology]] of amenorrhea is multifactorial and include [[hypothalamic]], [[pituitary]], [[thyroid]], [[adrenal]], [[ovarian]], [[uterine]], and [[vaginal]] [[ | **Primary amenorrhea is the absence of [[menstrual cycle]] by 16 years of age, in the presence of normal growth and secondary sexual characteristics. | ||
**Secondary amenorrhea reflects absence of [[menstrual cycle]] for at least 3 months in a woman with normal [[menstruation]] cycles in the past. | |||
*The [[pathophysiology]] of amenorrhea is multifactorial and include [[hypothalamic]], [[pituitary]], [[thyroid]], [[adrenal]], [[ovarian]], [[uterine]], and [[vaginal]] [[causes]]. | |||
==== Hypothalamic pathogenesis ==== | ==== Hypothalamic pathogenesis ==== | ||
*The most common cause of amenorrhea in adolescents | *The most common cause of amenorrhea in adolescents are [[hypothalamic]] disorders and is known as [[hypothalamic]] amenorrhea. | ||
* | *During the initial 2-3 years after the onset of [[menarche]], the [[Hypothalamic-pituitary-gonadal axis|HPO axis]] is still under development. Immature [[Hypothalamic-pituitary-gonadal axis|HPO axis]] may lead to [[Anovulatory cycle|anovulatory cycles]] which can cause abnormalities in [[menstrual cycles]]. | ||
*The most common cause of amenorrhea after 2-3 years of puberty | *The most common cause of amenorrhea after 2-3 years of onset of puberty include [[eating disorders]], excessive [[exercise]], [[medications]], and psychosocial stress.<ref name="pmid17230292">{{cite journal |vauthors=Wiksten-Almströmer M, Hirschberg AL, Hagenfeldt K |title=Menstrual disorders and associated factors among adolescent girls visiting a youth clinic |journal=Acta Obstet Gynecol Scand |volume=86 |issue=1 |pages=65–72 |year=2007 |pmid=17230292 |doi=10.1080/00016340601034970 |url=}}</ref><ref name="pmid11574516">{{cite journal |vauthors=Perkins RB, Hall JE, Martin KA |title=Aetiology, previous menstrual function and patterns of neuro-endocrine disturbance as prognostic indicators in hypothalamic amenorrhoea |journal=Hum. Reprod. |volume=16 |issue=10 |pages=2198–205 |year=2001 |pmid=11574516 |doi= |url=}}</ref> | ||
*[[Leptin]] plays an important role in [[energy]] consumption, body composition, food intake along with sexual maturation and [[Reproductive system|reproductive]] improvement. | *[[Leptin]] plays an important role in [[energy]] consumption, body composition, food intake along with sexual maturation and [[Reproductive system|reproductive]] improvement. | ||
**It is assumed that [[leptin]] | **It is assumed that [[leptin]] plays a role in the development of [[hypothalamic]] [[amenorrhea]]. | ||
**[[Leptin]] receptors are in close relationship with [[hypothalamus]] and it is postulated that leptin regulates [[GnRH]] production and secretion. | **[[Leptin]] receptors are in close relationship with [[hypothalamus]] and it is postulated that leptin regulates [[GnRH]] production and secretion. | ||
** | **Patients with [[anorexia nervosa]] or excessive [[exercise]] have amenorrhea from [[down-regulation]] of [[leptin]] receptors, which can be elevated by [[refeeding]].<ref name="pmid17060920">{{cite journal |vauthors=Hebebrand J, Muller TD, Holtkamp K, Herpertz-Dahlmann B |title=The role of leptin in anorexia nervosa: clinical implications |journal=Mol. Psychiatry |volume=12 |issue=1 |pages=23–35 |year=2007 |pmid=17060920 |doi=10.1038/sj.mp.4001909 |url=}}</ref><ref name="pmid8923829">{{cite journal |vauthors=Grinspoon S, Gulick T, Askari H, Landt M, Lee K, Anderson E, Ma Z, Vignati L, Bowsher R, Herzog D, Klibanski A |title=Serum leptin levels in women with anorexia nervosa |journal=J. Clin. Endocrinol. Metab. |volume=81 |issue=11 |pages=3861–3 |year=1996 |pmid=8923829 |doi=10.1210/jcem.81.11.8923829 |url=}}</ref><ref name="pmid15817868">{{cite journal |vauthors=Haas V, Onur S, Paul T, Nutzinger DO, Bosy-Westphal A, Hauer M, Brabant G, Klein H, Müller MJ |title=Leptin and body weight regulation in patients with anorexia nervosa before and during weight recovery |journal=Am. J. Clin. Nutr. |volume=81 |issue=4 |pages=889–96 |year=2005 |pmid=15817868 |doi= |url=}}</ref> | ||
**[[Leptin]] level in [[cachexic]] patients will increase after gaining appropriate weight in | **[[Leptin]] level in [[cachexic]] patients will increase after gaining appropriate weight in patients without amenorrhea, but will remain low in patients with amenorrhea.<ref name="pmid15240636">{{cite journal |vauthors=Misra M, Miller KK, Almazan C, Ramaswamy K, Aggarwal A, Herzog DB, Neubauer G, Breu J, Klibanski A |title=Hormonal and body composition predictors of soluble leptin receptor, leptin, and free leptin index in adolescent girls with anorexia nervosa and controls and relation to insulin sensitivity |journal=J. Clin. Endocrinol. Metab. |volume=89 |issue=7 |pages=3486–95 |year=2004 |pmid=15240636 |doi=10.1210/jc.2003-032251 |url=}}</ref><ref name="pmid10832726">{{cite journal |vauthors=Katzman DK, Golden NH, Neumark-Sztainer D, Yager J, Strober M |title=From prevention to prognosis: clinical research update on adolescent eating disorders |journal=Pediatr. Res. |volume=47 |issue=6 |pages=709–12 |year=2000 |pmid=10832726 |doi= |url=}}</ref> | ||
*It has been observed that [[leptin]] levels in amenorrheic athletes are low as compared to non-athletes women or athletes with regular [[menses]].<ref name="pmid10846016">{{cite journal |vauthors=Thong FS, McLean C, Graham TE |title=Plasma leptin in female athletes: relationship with body fat, reproductive, nutritional, and endocrine factors |journal=J. Appl. Physiol. |volume=88 |issue=6 |pages=2037–44 |year=2000 |pmid=10846016 |doi= |url=}}</ref><ref name="pmid10583427">{{cite journal |vauthors=Weimann E, Blum WF, Witzel C, Schwidergall S, Böhles HJ |title=Hypoleptinemia in female and male elite gymnasts |journal=Eur. J. Clin. Invest. |volume=29 |issue=10 |pages=853–60 |year=1999 |pmid=10583427 |doi= |url=}}</ref> | *It has been observed that [[leptin]] levels in amenorrheic athletes are low as compared to non-athletes women or athletes with regular [[menses]].<ref name="pmid10846016">{{cite journal |vauthors=Thong FS, McLean C, Graham TE |title=Plasma leptin in female athletes: relationship with body fat, reproductive, nutritional, and endocrine factors |journal=J. Appl. Physiol. |volume=88 |issue=6 |pages=2037–44 |year=2000 |pmid=10846016 |doi= |url=}}</ref><ref name="pmid10583427">{{cite journal |vauthors=Weimann E, Blum WF, Witzel C, Schwidergall S, Böhles HJ |title=Hypoleptinemia in female and male elite gymnasts |journal=Eur. J. Clin. Invest. |volume=29 |issue=10 |pages=853–60 |year=1999 |pmid=10583427 |doi= |url=}}</ref> | ||
*Administration of [[recombinant]] [[leptin]] for 3 months in women with conditions such as [[hypothalamic]] [[amenorrhea]], excessive [[exercises]] or [[weight loss]] has been associated with an increased level of [[LH]], [[FSH]], and [[estradiol]] | *Administration of [[recombinant]] [[leptin]] for 3 months in women with conditions such as [[hypothalamic]] [[amenorrhea]], excessive [[exercises]] or [[weight loss]] has been associated with an increased level of [[LH]], [[FSH]], and [[estradiol]] leading to [[Ovulation|ovulatory]] cycles.<ref name="WeltChan2004">{{cite journal|last1=Welt|first1=Corrine K.|last2=Chan|first2=Jean L.|last3=Bullen|first3=John|last4=Murphy|first4=Robyn|last5=Smith|first5=Patricia|last6=DePaoli|first6=Alex M.|last7=Karalis|first7=Aspasia|last8=Mantzoros|first8=Christos S.|title=Recombinant Human Leptin in Women with Hypothalamic Amenorrhea|journal=New England Journal of Medicine|volume=351|issue=10|year=2004|pages=987–997|issn=0028-4793|doi=10.1056/NEJMoa040388}}</ref> | ||
*[[Antipsychotic drugs]] and other medications that have inhibitory effect on [[Dopamine receptor D2|dopamine D2 receptor]] | *[[Antipsychotic drugs]] and other medications that have an inhibitory effect on [[Dopamine receptor D2|dopamine D2 receptor]] lead to an increased level of [[prolactin]]. Higher levels of [[prolactin]] suppress pulsatile [[GnRH]] secretion and block positive [[feedback]] of [[estradiol]] on [[hypothalamus]], leading to disruption of [[Hypothalamic-pituitary-gonadal axis|HPO axis]].<ref name="pmid12611781">{{cite journal |vauthors=Wieck A, Haddad PM |title=Antipsychotic-induced hyperprolactinaemia in women: pathophysiology, severity and consequences. Selective literature review |journal=Br J Psychiatry |volume=182 |issue= |pages=199–204 |year=2003 |pmid=12611781 |doi= |url=}}</ref> | ||
*[[Stress]] and strenuous activities | *[[Stress]] and strenuous activities like other [[metabolic]] or [[cardiovascular]] responses are regulated through [[Corticotropin releasing hormone|corticotropin releasing hormone (CRH)]], secreted by [[Paraventricular nucleus of hypothalamus|paraventricular nuclei of hypothalamus]]. [[CRH]] induce the release of [[β-endorphin]], an endogenous [[opioid]]. Both [[CRH]] and [[β-endorphin]] suppress [[GnRH]] release. On the other hand, [[glucocorticoids]] suppress [[LH]] production from [[pituitary]] and also [[estrogen]]/[[progesterone]] from [[ovaries]].<ref name="pmid9238254">{{cite journal |vauthors=Magiakou MA, Mastorakos G, Webster E, Chrousos GP |title=The hypothalamic-pituitary-adrenal axis and the female reproductive system |journal=Ann. N. Y. Acad. Sci. |volume=816 |issue= |pages=42–56 |year=1997 |pmid=9238254 |doi= |url=}}</ref> | ||
*[[Kallmann syndrome]], a [[genetic disorder]] caused by [[KAL1 gene|KAL gene]] [[mutation]], has disturbance in migration of [[olfactory nerves]] along with [[GnRH]] neurons. Lack of [[GnRH]] leads to absence of [[secondary sexual characteristics]] and amenorrhea.<ref name="pmid9793755">{{cite journal |vauthors=Seminara SB, Hayes FJ, Crowley WF |title=Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations |journal=Endocr. Rev. |volume=19 |issue=5 |pages=521–39 |year=1998 |pmid=9793755 |doi=10.1210/edrv.19.5.0344 |url=}}</ref> | *[[Kallmann syndrome]], a [[genetic disorder]] caused by [[KAL1 gene|KAL gene]] [[mutation]], has disturbance in migration of [[olfactory nerves]] along with [[GnRH]] neurons. Lack of [[GnRH]] leads to absence of [[secondary sexual characteristics]] and amenorrhea.<ref name="pmid9793755">{{cite journal |vauthors=Seminara SB, Hayes FJ, Crowley WF |title=Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations |journal=Endocr. Rev. |volume=19 |issue=5 |pages=521–39 |year=1998 |pmid=9793755 |doi=10.1210/edrv.19.5.0344 |url=}}</ref> | ||
==== Pituitary pathogenesis ==== | ==== Pituitary pathogenesis ==== | ||
* | * P[[prolactinoma|rolactinoma]] is one of the most common [[anterior pituitary]] [[tumors]]. [[Prolactinoma]] leads to [[Hyperprolactinemia|increased prolactin secretion]] and along with the [[Tumor|tumor's]] mass effect may cause suppression of [[Gonadotropin-releasing hormone|GnRH]]. | ||
* | * The second most common tumor in the [[suprasellar]] region is [[craniopharyngioma]]. The [[tumor]] leads to [[LH]] and [[FSH]] disturbances, which may cause amenorrhea.<ref name="pmid16543382">{{cite journal |vauthors=Karavitaki N, Cudlip S, Adams CB, Wass JA |title=Craniopharyngiomas |journal=Endocr. Rev. |volume=27 |issue=4 |pages=371–97 |year=2006 |pmid=16543382 |doi=10.1210/er.2006-0002 |url=}}</ref> | ||
==== Thyroid pathogenesis ==== | ==== Thyroid pathogenesis ==== | ||
| Line 55: | Line 59: | ||
==== Adrenal pathogenesis ==== | ==== Adrenal pathogenesis ==== | ||
* [[Congenital adrenal hyperplasia (CAH)]] is a group of [[genetic]] [[enzyme]] | * [[Congenital adrenal hyperplasia (CAH)]] is a group of [[genetic]] [[enzyme]] deficiencies in [[adrenal gland]]. Most common is the defect in [[21-hydroxylase|21-hydroxylase enzyme]], which leads to decrease in the level of [[aldosterone]] and [[cortisol]]. To overcome the [[hormone]] deficiencies, [[CRH]] production is increased by the [[hypothalamus]]. As described earlier, [[increased]] level of [[CRH]] may suppress [[GnRH]] and lead to amenorrhea.<ref name="SpeiserWhite2003">{{cite journal|last1=Speiser|first1=Phyllis W.|last2=White|first2=Perrin C.|title=Congenital Adrenal Hyperplasia|journal=New England Journal of Medicine|volume=349|issue=8|year=2003|pages=776–788|issn=0028-4793|doi=10.1056/NEJMra021561}}</ref> | ||
* [[Cushing syndrome]] has increased level of [[cortisol]], that can directly inhibit [[Hypothalamic-pituitary-gonadal axis|HPO axis]] and | * [[Cushing syndrome]] has an increased level of [[cortisol]], that can directly inhibit [[Hypothalamic-pituitary-gonadal axis|HPO axis]] and lead to amenorrhea. | ||
==== Ovarian pathogenesis ==== | ==== Ovarian pathogenesis ==== | ||
* [[Polycystic ovary syndrome| | * In [[Polycystic ovary syndrome|polycystic ovary syndrome (PCOS)]] [[insulin resistance]] leads to increased [[androgen]] production ([[insulin]] reduces the [[Sex hormone-binding globulin|SHBG]] circulating in [[plasma]], causing increased [[testosterone]]). In [[ovary|ovaries]], increased stimulation from [[GnRH]] leads to increased production of [[17-Hydroxyprogesterone|17-hydroxy progesterone]] and [[Cytochrome P450|cytochrome P450c17]] which promotes [[androgens]] [[biosynthesis]].<ref name="pmid9302378">{{cite journal |vauthors=Gilling-Smith C, Story H, Rogers V, Franks S |title=Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome |journal=Clin. Endocrinol. (Oxf) |volume=47 |issue=1 |pages=93–9 |year=1997 |pmid=9302378 |doi= |url=}}</ref><ref name="pmid7671850">{{cite journal |vauthors=Ehrmann DA, Barnes RB, Rosenfield RL |title=Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion |journal=Endocr. Rev. |volume=16 |issue=3 |pages=322–53 |year=1995 |pmid=7671850 |doi=10.1210/edrv-16-3-322 |url=}}</ref> Finally, the pulsatility of [[GnRH]] will be disrupted leading to amenorrhea .<ref name="pmid9467578">{{cite journal |vauthors=Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC |title=Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone |journal=J. Clin. Endocrinol. Metab. |volume=83 |issue=2 |pages=582–90 |year=1998 |pmid=9467578 |doi=10.1210/jcem.83.2.4604 |url=}}</ref> | ||
* [[Primary ovarian failure|Primary ovarian insufficiency]] | * [[Primary ovarian failure|Primary ovarian insufficiency]] is multifactorial and leads to [[ovarian failure]] and decrease in [[estrogen]] which leads to amenorrhea. (In [[galactosemia]], it is assumed that [[galactose]] and its metabolites are toxic to [[ovarian]] tissue.)<ref name="pmid6782485">{{cite journal |vauthors=Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R |title=Hypergonadotropic hypogonadism in female patients with galactosemia |journal=N. Engl. J. Med. |volume=304 |issue=17 |pages=994–8 |year=1981 |pmid=6782485 |doi=10.1056/NEJM198104233041702 |url=}}</ref> | ||
==== Uterine pathogenesis ==== | ==== Uterine pathogenesis ==== | ||
* The main [[pathogenesis]] of amenorrhea in [[androgen insensitivity syndrome]] is absence of [[uterus]]. The patient is [[Genotype| | * The main [[pathogenesis]] of amenorrhea in [[androgen insensitivity syndrome]] is the absence of [[uterus]]. The patient is [[Genotype|genotypically]] male, 46 XY; but with absent [[sexual characteristics]] due to lack of functional effect of [[Androgen|androgen hormones]] on their receptors. | ||
* One of the acquired conditions that can lead to amenorrhea is [[Asherman syndrome]]. The | * One of the acquired conditions that can lead to amenorrhea is [[Asherman syndrome]]. The basis of [[Asherman syndrome]] is any condition that can alter the normal histology of [[endometrium]], such as [[scarring]] from [[surgical procedure]] or [[adhesion]] from severe [[infection]]. | ||
* [[Mayer-Rokitansky-Hauser syndrome|Mayer-Rokitansky-Kuster-Hauser syndrome]] is complete [[agenesis]] of [[uterine]], blind ended [[vagina]]. The lack of [[uterine]] and [[endometrium]] is the main [[pathogenesis]] of amenorrhea. The main reason of [[Uterine atresia|uterine agenesis]] is overactivation of [[Antimullerian hormone|anti-mullerian hormone]] in [[embryogenesis]] period.<ref name="pmid4020785">{{cite journal |vauthors=Varner RE, Younger JB, Blackwell RE |title=Müllerian dysgenesis |journal=J Reprod Med |volume=30 |issue=6 |pages=443–50 |year=1985 |pmid=4020785 |doi= |url=}}</ref> [[Cervical]] [[agenesis]] also follow the similar process. | * [[Mayer-Rokitansky-Hauser syndrome|Mayer-Rokitansky-Kuster-Hauser syndrome]] is complete [[agenesis]] of [[uterine]], blind ended [[vagina]]. The lack of [[uterine]] and [[endometrium]] is the main [[pathogenesis]] of amenorrhea. The main reason of [[Uterine atresia|uterine agenesis]] is overactivation of [[Antimullerian hormone|anti-mullerian hormone]] in [[embryogenesis]] period.<ref name="pmid4020785">{{cite journal |vauthors=Varner RE, Younger JB, Blackwell RE |title=Müllerian dysgenesis |journal=J Reprod Med |volume=30 |issue=6 |pages=443–50 |year=1985 |pmid=4020785 |doi= |url=}}</ref> [[Cervical]] [[agenesis]] also follow the similar process. | ||
* [[Imperforate hymen|Imperforated hymen]], [[Vaginal septum|transverse vaginal septum]], and [[vaginal agenesis]] are other [[anatomical]] disorders of [[female reproductive system]] that can lead to amenorrhea.<ref name="pmid12758224">{{cite journal |vauthors=Edmonds DK |title=Congenital malformations of the genital tract and their management |journal=Best Pract Res Clin Obstet Gynaecol |volume=17 |issue=1 |pages=19–40 |year=2003 |pmid=12758224 |doi= |url=}}</ref> | * [[Imperforate hymen|Imperforated hymen]], [[Vaginal septum|transverse vaginal septum]], and [[vaginal agenesis]] are other [[anatomical]] disorders of [[female reproductive system]] that can lead to amenorrhea.<ref name="pmid12758224">{{cite journal |vauthors=Edmonds DK |title=Congenital malformations of the genital tract and their management |journal=Best Pract Res Clin Obstet Gynaecol |volume=17 |issue=1 |pages=19–40 |year=2003 |pmid=12758224 |doi= |url=}}</ref> | ||
| Line 70: | Line 74: | ||
==Genetics== | ==Genetics== | ||
=== The major genes in amenorrhea === | === The major genes in amenorrhea === | ||
{| class="wikitable" | {| class="wikitable" | ||
!Groups | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Groups | ||
!Gene | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Gene | ||
!Other name(s) | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Other name(s) | ||
!OMIM number | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |OMIM number | ||
!Chromosome | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Chromosome | ||
!Function | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Function | ||
!Other related disorders | ! style="background:#4479BA; color: #FFFFFF;" align="center" + |Other related disorders | ||
|- | |- | ||
| rowspan="15" |'''Kallmann syndrome''' | | rowspan="15" style="background:#DCDCDC;" align="center" + |'''Kallmann syndrome''' | ||
and | and | ||
'''Isolated hypogonadotropic hypogonadism'''<ref name="BonomiLibri2011">{{cite journal|last1=Bonomi|first1=Marco|last2=Libri|first2=Domenico Vladimiro|last3=Guizzardi|first3=Fabiana|last4=Guarducci|first4=Elena|last5=Maiolo|first5=Elisabetta|last6=Pignatti|first6=Elisa|last7=Asci|first7=Roberta|last8=Persani|first8=Luca|title=New understandings of the genetic basis of isolated idiopathic central hypogonadism|journal=Asian Journal of Andrology|volume=14|issue=1|year=2011|pages=49–56|issn=1008-682X|doi=10.1038/aja.2011.68}}</ref> | '''Isolated hypogonadotropic hypogonadism'''<ref name="BonomiLibri2011">{{cite journal|last1=Bonomi|first1=Marco|last2=Libri|first2=Domenico Vladimiro|last3=Guizzardi|first3=Fabiana|last4=Guarducci|first4=Elena|last5=Maiolo|first5=Elisabetta|last6=Pignatti|first6=Elisa|last7=Asci|first7=Roberta|last8=Persani|first8=Luca|title=New understandings of the genetic basis of isolated idiopathic central hypogonadism|journal=Asian Journal of Andrology|volume=14|issue=1|year=2011|pages=49–56|issn=1008-682X|doi=10.1038/aja.2011.68}}</ref> | ||
|'''KAL1''' | | style="background:#DCDCDC;" align="center" + |'''KAL1''' | ||
|[[KAL1 gene|KAL1]], [[anosmin-1]] | |[[KAL1 gene|KAL1]], [[anosmin-1]] | ||
|308700 | |308700 | ||
| Line 102: | Line 104: | ||
* [[Cerebellar ataxia]] | * [[Cerebellar ataxia]] | ||
|- | |- | ||
|'''FGFR1''' | | style="background:#DCDCDC;" align="center" + |'''FGFR1''' | ||
|KAL2 | |KAL2 | ||
|136350 | |136350 | ||
| Line 117: | Line 119: | ||
* Bimanual [[synkinesis]] | * Bimanual [[synkinesis]] | ||
|- | |- | ||
|'''PROKR2''' | | style="background:#DCDCDC;" align="center" + |'''PROKR2''' | ||
|KAL3 | |KAL3 | ||
|607123 | |607123 | ||
| Line 133: | Line 135: | ||
* [[Epilepsy]] | * [[Epilepsy]] | ||
|- | |- | ||
|'''PROK2''' | | style="background:#DCDCDC;" align="center" + |'''PROK2''' | ||
|KAL4 | |KAL4 | ||
|607002 | |607002 | ||
|3p21.1 | |3p21.1 | ||
|- | |- | ||
|'''CHD7''' | | style="background:#DCDCDC;" align="center" + |'''CHD7''' | ||
|KAL5 | |KAL5 | ||
|608892 | |608892 | ||
| Line 153: | Line 155: | ||
** [[Ear|'''E'''ar]] anomalies | ** [[Ear|'''E'''ar]] anomalies | ||
|- | |- | ||
|'''FGF8''' | | style="background:#DCDCDC;" align="center" + |'''FGF8''' | ||
|KAL6 | |KAL6 | ||
|600483 | |600483 | ||
| Line 168: | Line 170: | ||
* [[Cerebellar]] developmental abnormalities | * [[Cerebellar]] developmental abnormalities | ||
|- | |- | ||
|'''GPR54''' | | style="background:#DCDCDC;" align="center" + |'''GPR54''' | ||
|KISS1R | |KISS1R | ||
|604161 | |604161 | ||
| Line 176: | Line 178: | ||
| - | | - | ||
|- | |- | ||
|'''KISS1''' | | style="background:#DCDCDC;" align="center" + |'''KISS1''' | ||
|KISS1, kisspeptin1 | |KISS1, kisspeptin1 | ||
|603286 | |603286 | ||
| Line 184: | Line 186: | ||
| - | | - | ||
|- | |- | ||
|'''HS6ST1''' | | style="background:#DCDCDC;" align="center" + |'''HS6ST1''' | ||
| - | | - | ||
|604846 | |604846 | ||
| Line 194: | Line 196: | ||
| - | | - | ||
|- | |- | ||
|'''TAC3''' | | style="background:#DCDCDC;" align="center" + |'''TAC3''' | ||
|NKB | |NKB | ||
|162330 | |162330 | ||
| Line 207: | Line 209: | ||
* [[Cryptorchidism]] | * [[Cryptorchidism]] | ||
|- | |- | ||
|'''TACR3''' | | style="background:#DCDCDC;" align="center" + |'''TACR3''' | ||
|NK3R | |NK3R | ||
|152332 | |152332 | ||
|4q25 | |4q25 | ||
|- | |- | ||
|'''GnRH1''' | | style="background:#DCDCDC;" align="center" + |'''GnRH1''' | ||
| - | | - | ||
|152760 | |152760 | ||
| Line 221: | Line 223: | ||
* [[Teeth]] abnormal [[maturation]] and biomineralization | * [[Teeth]] abnormal [[maturation]] and biomineralization | ||
|- | |- | ||
|'''GnRHR''' | | style="background:#DCDCDC;" align="center" + |'''GnRHR''' | ||
| - | | - | ||
|138850 | |138850 | ||
| Line 233: | Line 235: | ||
* Failure to impact from exogenous [[GnRH]] | * Failure to impact from exogenous [[GnRH]] | ||
|- | |- | ||
|'''NELF''' | | style="background:#DCDCDC;" align="center" + |'''NELF''' | ||
| - | | - | ||
|608137 | |608137 | ||
|''9q34.3'' | |''9q34.3'' | ||
| | | | ||
* | * Modulating [[neuron]] migration in developmental process | ||
* | * [[Olfactory]] axons and also [[GnRH]] [[neurons]] functions | ||
| - | | - | ||
|- | |- | ||
|'''EBF2''' | | style="background:#DCDCDC;" align="center" + |'''EBF2''' | ||
| - | | - | ||
|609934 | |609934 | ||
| Line 250: | Line 252: | ||
| - | | - | ||
|- | |- | ||
| rowspan="5" |'''HPG axis development''' | | rowspan="5" style="background:#DCDCDC;" align="center" + |'''HPG axis development''' | ||
|'''DAX1''' | | style="background:#DCDCDC;" align="center" + |'''DAX1''' | ||
|NR0B | |NR0B | ||
|300473 | |300473 | ||
| Line 262: | Line 264: | ||
* Congenital [[Adrenal cortex insufficiency|adrenal cortex hypoplasia]] | * Congenital [[Adrenal cortex insufficiency|adrenal cortex hypoplasia]] | ||
|- | |- | ||
|'''SF-1''' | | style="background:#DCDCDC;" align="center" + |'''SF-1''' | ||
|NR5A1 | |NR5A1 | ||
|184757 | |184757 | ||
| Line 275: | Line 277: | ||
* [[Hypospadias]] | * [[Hypospadias]] | ||
|- | |- | ||
|'''HESX-1''' | | style="background:#DCDCDC;" align="center" + |'''HESX-1''' | ||
|RPX | |RPX | ||
|601802 | |601802 | ||
|''3p14.3'' | |''3p14.3'' | ||
| | | | ||
* | * [[Pituitary]] development | ||
* | * Midfacial differentiation | ||
* | * [[Mutation]] may lead to [[pituitary]] [[hypoplasia]] | ||
| | | | ||
* Septooptic dysplasia | * Septooptic dysplasia | ||
| Line 292: | Line 294: | ||
* Abnormalities in the [[corpus callosum]], [[hippocampus]], and [[septum pellucidum]] | * Abnormalities in the [[corpus callosum]], [[hippocampus]], and [[septum pellucidum]] | ||
|- | |- | ||
|'''LHX3''' | | style="background:#DCDCDC;" align="center" + |'''LHX3''' | ||
|LIM3 | |LIM3 | ||
|600577 | |600577 | ||
| Line 305: | Line 307: | ||
* Skeletal abnormalities | * Skeletal abnormalities | ||
|- | |- | ||
|'''PROP-1''' | | style="background:#DCDCDC;" align="center" + |'''PROP-1''' | ||
| - | | - | ||
|601538 | |601538 | ||
|''5q35.3'' | |''5q35.3'' | ||
| | | | ||
* | * Developing anterior [[pituitary gland]] | ||
* | * [[Gonadotrophs]] | ||
* | * [[Thyrotrophs|Tthyrotrophs]] | ||
* | * [[Somatotrophs]] | ||
* | * [[Lactotrophs|Lactotrophs]] | ||
* Low [[LH]] and [[FSH]] delay the [[puberty]] | * Low [[LH]] and [[FSH]] delay the [[puberty]] | ||
| | | | ||
| Line 321: | Line 323: | ||
* [[Libido]]/[[Lactation]] problems | * [[Libido]]/[[Lactation]] problems | ||
|- | |- | ||
| rowspan="3" |'''Obesity related''' | | rowspan="3" style="background:#DCDCDC;" align="center" + |'''Obesity related''' | ||
'''hypogonadotropic hypogonadism''' | '''hypogonadotropic hypogonadism''' | ||
|'''LEP''' | | style="background:#DCDCDC;" align="center" + |'''LEP''' | ||
|OB | |OB | ||
|164160 | |164160 | ||
|''7q32.1'' | |''7q32.1'' | ||
| rowspan="2" | | | rowspan="2" | | ||
* | * Modulation of [[body weight]] | ||
* Beginning the [[puberty]] | * Beginning the [[puberty]] | ||
* [[Recombinant]] [[leptin]] injection in female mice result in [[puberty]] | * [[Recombinant]] [[leptin]] injection in female mice result in [[puberty]] | ||
| Line 338: | Line 340: | ||
* [[Immune]] or [[inflammatory response]] disorders | * [[Immune]] or [[inflammatory response]] disorders | ||
|- | |- | ||
|'''LEPR''' | | style="background:#DCDCDC;" align="center" + |'''LEPR''' | ||
|OBR | |OBR | ||
|601007 | |601007 | ||
|''1p31.3'' | |''1p31.3'' | ||
|- | |- | ||
|'''PC1''' | | style="background:#DCDCDC;" align="center" + |'''PC1''' | ||
|NEC1 | |NEC1 | ||
|162150 | |162150 | ||
|''5q15'' | |''5q15'' | ||
| | | | ||
* | * Regulates [[neuroendocrine]] pathway | ||
* [[Proopiomelanocortin]] (POMC) cleavage | * [[Proopiomelanocortin]] (POMC) cleavage | ||
* Processing [[proinsulin]] and [[proglucagon]] in [[pancreas]]. | * Processing [[proinsulin]] and [[proglucagon]] in [[pancreas]]. | ||
| Line 355: | Line 357: | ||
* Abnormal glucose [[homeostasis]] | * Abnormal glucose [[homeostasis]] | ||
* [[Hypocortisolism]] | * [[Hypocortisolism]] | ||
* Elevated plasma [[proinsulin]], and also [[POMC]] | * Elevated plasma [[proinsulin]], and also [[POMC]] | ||
|- | |||
| colspan="7" style="background:#DCDCDC;" + | | |||
<span style="font-size:85%">'''Abbreviations (alphabetic):'''<br> | |||
'''CHD7:''' Chromodomain [[helicase]] DNA-binding protein 7 gene, '''DAX1:''' DSS-AHC on the [[X-chromosome]] 1, '''EBF2:''' Early [[B-cell]] factor 2 gene, '''FGF8:''' [[Fibroblast growth factor 8]] gene, '''FGFR1:''' [[Fibroblast growth factor receptor 1]] gene, '''FSH:''' [[Follicle stimulating hormone]], '''GnRH:''' [[Gonadotropin releasing hormone]], '''GnRH1:''' [[Gonadotropin releasing hormone]] 1 gene, '''GnRHR:''' [[Gonadotropin releasing hormone]] receptor gene, '''GPR54:''' [[G protein-coupled receptor|G protein-coupled receptor-54]] gene, '''HESX-1:''' [[Homeobox]] gene 1, '''HPG axis:''' Hypothalamus-pituitary-gonadal axis, '''HS6ST1:''' [[Heparan sulfate]] 6-O-sulphotransferase 1 gene, '''KAL1:''' [[Kallman syndrome|Kallmann syndrome]] 1 gene, '''LEP:''' [[Leptin]] gene''', LEPR:''' [[Leptin receptor]] gene''', LH:''' [[Luteinizing hormone]], '''LHX3:''' LIM [[homeobox]] gene 3''', NEC1:''' [[Neuroendocrine]] convertase 1, '''NELF:''' Nasal embryonic LH-releasing hormone factor gene, '''NK3R:''' [[Neurokinin]] 3 receptor gene, '''NKB:''' [[Neurokinin B]] gene, '''NR0B:''' [[Nuclear receptor]] 0B, '''NR5A1:''' [[Nuclear receptor]] 5A1, '''OMIM:''' [[Online Mendelian Inheritance in Man]], '''PC1:''' [[Proprotein]] convertase 1''', PROK2 :''' [[Prokineticin]] 2 gene, '''PROKR2:''' [[Prokineticin]] 2 receptor gene, '''PROP-1:''' [[PROP]] paired-like homeobox 1, '''RPX:''' [[Rathke pouch]] homeobox, '''SF-1:''' [[Steroidogenic]] factor 1, '''TAC3:''' [[Tachykinin]] 3 gene,'''TACR3:''' [[Tachykinin]] 3 receptor gene, | |||
</span> | |||
|} | |} | ||
=== Kisspeptin system (KISS1R and KISS1) === | === Kisspeptin system (KISS1R and KISS1) === | ||
* The GPR54 [[gene]], also called KISS1R, with [[Online Mendelian Inheritance in Man|Online Mendelian Inheritance in Man (OMIM)]] number of 604161 is on chromosome 19p13.3. The KISS1 gene, also | * The GPR54 [[gene]], also called KISS1R, with [[Online Mendelian Inheritance in Man|Online Mendelian Inheritance in Man (OMIM)]] number of 604161 is on chromosome 19p13.3. The KISS1 gene, also known as [[Kisspeptin|kisspeptin1]], with [[OMIM]] number of 603286 is on [[chromosome]] 1q32. | ||
* | * [[Kisspeptin]] and related [[G-protein coupled receptor]] (KISS1R or GPR54) have key roles in the regulation of [[GnRH]] secretion. The [[GnRH]] secretion has to be pulsatile to stimulate [[gonadotropins]]. [[Kisspeptin|Kisspeptins]] are encoded by KISS1 gene, [[neuropeptides]] secreted from [[hypothalamus]] nuclei. It has been observed that patients with idiopathic [[hypogonadotropic hypogonadism]] have KISS1 receptor (GPR54) inactivating [[gene]] [[mutations]].<ref name="pmid12944565">{{cite journal |vauthors=de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E |title=Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54 |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=100 |issue=19 |pages=10972–6 |year=2003 |pmid=12944565 |pmc=196911 |doi=10.1073/pnas.1834399100 |url=}}</ref><ref name="SeminaraMessager2003">{{cite journal|last1=Seminara|first1=Stephanie B.|last2=Messager|first2=Sophie|last3=Chatzidaki|first3=Emmanouella E.|last4=Thresher|first4=Rosemary R.|last5=Acierno|first5=James S.|last6=Shagoury|first6=Jenna K.|last7=Bo-Abbas|first7=Yousef|last8=Kuohung|first8=Wendy|last9=Schwinof|first9=Kristine M.|last10=Hendrick|first10=Alan G.|last11=Zahn|first11=Dirk|last12=Dixon|first12=John|last13=Kaiser|first13=Ursula B.|last14=Slaugenhaupt|first14=Susan A.|last15=Gusella|first15=James F.|last16=O'Rahilly|first16=Stephen|last17=Carlton|first17=Mark B.L.|last18=Crowley|first18=William F.|last19=Aparicio|first19=Samuel A.J.R.|last20=Colledge|first20=William H.|title=TheGPR54Gene as a Regulator of Puberty|journal=New England Journal of Medicine|volume=349|issue=17|year=2003|pages=1614–1627|issn=0028-4793|doi=10.1056/NEJMoa035322}}</ref> | ||
* By the time of [[puberty]], the KISS1 genes become activated through [[neuroanatomical]] and functional changes from environmental triggers, critical for [[brain]] sexual [[maturation]] and HPG activation with pulsatile [[GnRH]].<ref name="pmid23015158">{{cite journal| author=Kaur KK, Allahbadia G, Singh M| title=Kisspeptins in human reproduction-future therapeutic potential. | journal=J Assist Reprod Genet | year= 2012 | volume= 29 | issue= 10 | pages= 999-1011 | pmid=23015158 | doi=10.1007/s10815-012-9856-1 | pmc=3492584 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=23015158 }}</ref> | * By the time of onset of [[puberty]], the KISS1 genes become activated through [[neuroanatomical]] and functional changes from environmental triggers, critical for [[brain]] sexual [[maturation]] and hypothalamic–pituitary–gonadal axis (HPG axis) activation with pulsatile [[GnRH]].<ref name="pmid23015158">{{cite journal| author=Kaur KK, Allahbadia G, Singh M| title=Kisspeptins in human reproduction-future therapeutic potential. | journal=J Assist Reprod Genet | year= 2012 | volume= 29 | issue= 10 | pages= 999-1011 | pmid=23015158 | doi=10.1007/s10815-012-9856-1 | pmc=3492584 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=23015158 }}</ref> | ||
* Along HPG axis [[neurons]], [[gamma-aminobutyric acid]] is [[inhibitory]] and [[glutamate]] is [[Excitatory neurotransmitter|excitatory neurotransmitters]]. In related KNDy [[neurons]] in [[arcuate nucleus]], the materials secreted are included [[kisspeptin]], [[neurokinin B]], and [[dynorphin A]]. Before | * Along HPG axis [[neurons]], [[gamma-aminobutyric acid]] is [[inhibitory]] and [[glutamate]] is [[Excitatory neurotransmitter|excitatory neurotransmitters]]. In related KNDy [[neurons]] in [[arcuate nucleus]], the materials secreted are included [[kisspeptin]], [[neurokinin B]], and [[dynorphin A]]. Before [[puberty]] begins, inhibitory [[dynorphin A]] is the dominant element; decreased by stimulatory effect of [[neurokinin B]], when [[puberty]] started. Conclusively, [[kisspeptin]] and [[GnRH]]/[[LH]] are increased.<ref name="UenoyamaTsukamura2014">{{cite journal|last1=Uenoyama|first1=Yoshihisa|last2=Tsukamura|first2=Hiroko|last3=Maeda|first3=Kei-ichiro|title=KNDy neuron as a gatekeeper of puberty onset|journal=Journal of Obstetrics and Gynaecology Research|volume=40|issue=6|year=2014|pages=1518–1526|issn=13418076|doi=10.1111/jog.12398}}</ref> | ||
=== Kallmann syndrome 1 (KAL1) === | === Kallmann syndrome 1 (KAL1) === | ||
* The [[KAL1 gene|KAL1]] [[gene]], also called [[anosmin-1]], with [[OMIM]] number of 308700 is on [[chromosome]] Xp22.3, | * The [[KAL1 gene|KAL1]] [[gene]], also called [[anosmin-1]], with [[OMIM]] number of 308700 is on [[chromosome]] Xp22.3, and encodes an [[Extracellular matrix protein|extracellular matrix glycoprotein]]. | ||
* [[Anosmin-1]] is expressed at five weeks of [[gestation]] in [[forebrain]] | * [[Anosmin-1]] is expressed at five weeks of [[gestation]] in [[forebrain]] near [[olfactory bulbs]] and stimulate the [[afferent fibers]] projections around it.<ref name="pmid10340754">{{cite journal |vauthors=Hardelin JP, Julliard AK, Moniot B, Soussi-Yanicostas N, Verney C, Schwanzel-Fukuda M, Ayer-Le Lievre C, Petit C |title=Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome |journal=Dev. Dyn. |volume=215 |issue=1 |pages=26–44 |year=1999 |pmid=10340754 |doi=10.1002/(SICI)1097-0177(199905)215:1<26::AID-DVDY4>3.0.CO;2-D |url=}}</ref> | ||
* [[X-linked]] [[Kallman syndrome|Kallmann syndrome]] is directly associated with [[KAL1 gene|KAL1]] deletion. It is assumed to result in an absence of [[Olfactory system|olfactory fibers]] along with | * [[X-linked]] [[Kallman syndrome|Kallmann syndrome]] is directly associated with [[KAL1 gene|KAL1]] deletion. It is assumed to result in an absence of [[Olfactory system|olfactory fibers]] along with disrupted migration of [[GnRH]] [[neurons]], that are supposed to from migrated [[olfactory placode]].<ref name="pmid2687610">{{cite journal |vauthors=Schwanzel-Fukuda M, Bick D, Pfaff DW |title=Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome |journal=Brain Res. Mol. Brain Res. |volume=6 |issue=4 |pages=311–26 |year=1989 |pmid=2687610 |doi= |url=}}</ref> | ||
* Male patient with [[KAL1 gene|KAL1]] [[mutation]] would have central [[hypogonadism]] and [[anosmia]]/[[hyposmia]]. Additionally, the more [[diseases]] are assumed to relate with [[KAL1 gene]], such as midline [[facial]] defects ([[cleft lip]] and/or [[cleft palate]]), short [[metacarpals]], [[renal agenesis]], [[sensorineural hearing loss]], bimanual [[synkinesis]], [[oculomotor]] abnormalities, and [[cerebellar ataxia]].<ref name="pmid17624596">{{cite journal |vauthors=Trarbach EB, Silveira LG, Latronico AC |title=Genetic insights into human isolated gonadotropin deficiency |journal=Pituitary |volume=10 |issue=4 |pages=381–91 |year=2007 |pmid=17624596 |doi=10.1007/s11102-007-0061-7 |url=}}</ref> | * Male patient with [[KAL1 gene|KAL1]] [[mutation]] would have central [[hypogonadism]] and [[anosmia]]/[[hyposmia]]. Additionally, the more [[diseases]] are assumed to relate with [[KAL1 gene]], such as midline [[facial]] defects ([[cleft lip]] and/or [[cleft palate]]), short [[metacarpals]], [[renal agenesis]], [[sensorineural hearing loss]], bimanual [[synkinesis]], [[oculomotor]] abnormalities, and [[cerebellar ataxia]].<ref name="pmid17624596">{{cite journal |vauthors=Trarbach EB, Silveira LG, Latronico AC |title=Genetic insights into human isolated gonadotropin deficiency |journal=Pituitary |volume=10 |issue=4 |pages=381–91 |year=2007 |pmid=17624596 |doi=10.1007/s11102-007-0061-7 |url=}}</ref> | ||
=== Fibroblast growth factor receptor 1 and fibroblast growth factor 8 (FGFR1 and FGF8) === | === Fibroblast growth factor receptor 1 and fibroblast growth factor 8 (FGFR1 and FGF8) === | ||
* The [[FGFR1]] [[gene]], also called KAL2, with [[OMIM]] number of 136350 is on [[chromosome]] 8q12, encode a receptor [[Tyrosine kinase|tyrosine kinase protein]]. The [[FGF8]] gene, also called KAL6, is on [[chromosome]] 10q24. | * The [[FGFR1]] [[gene]], also called KAL2, with [[OMIM]] number of 136350 is on [[chromosome]] 8q12, encode a receptor [[Tyrosine kinase|tyrosine kinase protein]]. The [[FGF8]] gene, also called KAL6, is on [[chromosome]] 10q24. | ||
* [[FGFR1]] pathway is assumed to be the main role in [[embryogenesis]], [[homeostasis]], and [[wound healing]]. [[FGF8]] critical role in primary generation of [[neural tissue]] | * [[FGFR1]] pathway is assumed to be the main role in [[embryogenesis]], [[homeostasis]], and [[wound healing]]. [[FGF8]] plays a critical role in the primary generation of [[neural tissue]].<ref name="pmid15548653">{{cite journal |vauthors=González-Martínez D, Kim SH, Hu Y, Guimond S, Schofield J, Winyard P, Vannelli GB, Turnbull J, Bouloux PM |title=Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism |journal=J. Neurosci. |volume=24 |issue=46 |pages=10384–92 |year=2004 |pmid=15548653 |doi=10.1523/JNEUROSCI.3400-04.2004 |url=}}</ref> | ||
* On the other hand, interaction between [[FGFR1]], [[FGF8]], and [[heparan sulfate]] helps [[olfactory bulb]] to become differentiated and developed | * On the other hand, interaction between [[FGFR1]], [[FGF8]], and [[heparan sulfate]] helps the [[olfactory bulb]] to become differentiated and developed and also facilitates [[GnRH]] [[neurons]] in migration and function.<ref name="pmid12571102">{{cite journal |vauthors=Hébert JM, Lin M, Partanen J, Rossant J, McConnell SK |title=FGF signaling through FGFR1 is required for olfactory bulb morphogenesis |journal=Development |volume=130 |issue=6 |pages=1101–11 |year=2003 |pmid=12571102 |doi= |url=}}</ref> | ||
* Dominant [[deletion mutation]] of [[FGFR1]] gene is | * Dominant [[deletion mutation]] of [[FGFR1]] gene is associated with a 30% decrease in [[hypothalamic]] [[GnRH]] [[neurons]].<ref name="pmid15459253">{{cite journal |vauthors=Tsai PS, Moenter SM, Postigo HR, El Majdoubi M, Pak TR, Gill JC, Paruthiyil S, Werner S, Weiner RI |title=Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population |journal=Mol. Endocrinol. |volume=19 |issue=1 |pages=225–36 |year=2005 |pmid=15459253 |doi=10.1210/me.2004-0330 |url=}}</ref> Other defects related to [[FGFR1]] includes [[cleft palate]] or [[Cleft lip|lip]], dental [[agenesis]] and bimanual [[synkinesis]].<ref name="pmid17624596" /> | ||
=== Heparan sulfate 6-O-sulphotransferase 1 (HS6ST1) === | === Heparan sulfate 6-O-sulphotransferase 1 (HS6ST1) === | ||
* The HS6ST1 [[gene]] with [[OMIM]] number of 604846 is on [[chromosome]] 2q21 | * The HS6ST1 [[gene]] with [[OMIM]] number of 604846 is on [[chromosome]] 2q21 has been found to be mutated in [[hypogonadism]].<ref name="pmid21700882">{{cite journal |vauthors=Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Seminara SB, Hughes V, Van Vliet G, Van Uum S, Crowley WF, Habuchi H, Kimata K, Pitteloud N, Bülow HE |title=Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=108 |issue=28 |pages=11524–9 |year=2011 |pmid=21700882 |pmc=3136273 |doi=10.1073/pnas.1102284108 |url=}}</ref> | ||
* The modifications of [[heparan sulfate]] [[polysaccharides]] in [[extracellular matrix]] have some | * The modifications of [[heparan sulfate]] [[polysaccharides]] in [[extracellular matrix]] have some role in [[FGFR]]-[[FGF1|FGF]] and also [[Anosmin-1|anosmin1]]-[[cell membrane]] interactions.<ref name="pmid15096041">{{cite journal |vauthors=Ibrahimi OA, Zhang F, Hrstka SC, Mohammadi M, Linhardt RJ |title=Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly |journal=Biochemistry |volume=43 |issue=16 |pages=4724–30 |year=2004 |pmid=15096041 |doi=10.1021/bi0352320 |url=}}</ref><ref name="pmid16677626">{{cite journal |vauthors=Hudson ML, Kinnunen T, Cinar HN, Chisholm AD |title=C. elegans Kallmann syndrome protein KAL-1 interacts with syndecan and glypican to regulate neuronal cell migrations |journal=Dev. Biol. |volume=294 |issue=2 |pages=352–65 |year=2006 |pmid=16677626 |doi=10.1016/j.ydbio.2006.02.036 |url=}}</ref> | ||
* This [[gene]] | * This [[gene]] may be mutated in both [[Kallman syndrome|Kallmann syndrome]] and idiopathic [[hypogonadism]] resulting in various courses, disruption pf frequency of [[GnRH]] secretion, and/or [[GnRH]] deficiencies.<ref name="pmid21700882" /> | ||
=== Prokineticin 2 and prokineticin 2 receptor (PROK2 and PROKR2) === | === Prokineticin 2 and prokineticin 2 receptor (PROK2 and PROKR2) === | ||
* The [[Prokineticin|PROK2]] and [[Prokineticin receptor 2|PROKR2]] [[genes]], also called KAL4 and KAL3, with [[OMIM]] numbers of 607002 and 607123 are on [[chromosomes]] 3p21.1 and 20p13, respectively. They are believed to be cause of [[Kallman syndrome|Kallmann syndrome]]. | * The [[Prokineticin|PROK2]] and [[Prokineticin receptor 2|PROKR2]] [[genes]], also called KAL4 and KAL3, with [[OMIM]] numbers of 607002 and 607123 are on [[chromosomes]] 3p21.1 and 20p13, respectively. They are believed to be a cause of [[Kallman syndrome|Kallmann syndrome]]. | ||
* [[Prokineticin|PROKR2]] is a [[G protein coupled receptor|G protein coupled receptor (GPCR)]], has a major role in [[olfactory bulb]] development; | * [[Prokineticin|PROKR2]] is a [[G protein coupled receptor|G protein coupled receptor (GPCR)]], has a major role in [[olfactory bulb]] development; and its [[mutation]] may lead to severe [[gonadal]] [[atrophy]].<ref name="pmid16537498">{{cite journal |vauthors=Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y |title=Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2 |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=103 |issue=11 |pages=4140–5 |year=2006 |pmid=16537498 |pmc=1449660 |doi=10.1073/pnas.0508881103 |url=}}</ref> | ||
* In [[prokineticin]] system, there are two receptors ([[Prokineticin receptor 1|PROKR1]] and [[Prokineticin receptor 2|PROKR2]]) and two [[ligands]] ([[Prokineticin|PROK1]] and [[Prokineticin|PROK2]]). [[Prokineticin|PROK1]] and its receptor ([[Prokineticin receptor 1|PROKR1]]) have some | * In [[prokineticin]] system, there are two receptors ([[Prokineticin receptor 1|PROKR1]] and [[Prokineticin receptor 2|PROKR2]]) and two [[ligands]] ([[Prokineticin|PROK1]] and [[Prokineticin|PROK2]]). [[Prokineticin|PROK1]] and its receptor ([[Prokineticin receptor 1|PROKR1]]) have some role in [[gastrointestinal]] system [[motility]]. [[Prokineticin|PROK2]] and [[Prokineticin receptor 2|PROKR2]] are parts of [[neuroendocrine system]], located in [[arcuate nucleus]], [[olfactory tract]], and [[suprachiasmatic nucleus]].<ref name="pmid11259612">{{cite journal |vauthors=Li M, Bullock CM, Knauer DJ, Ehlert FJ, Zhou QY |title=Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle |journal=Mol. Pharmacol. |volume=59 |issue=4 |pages=692–8 |year=2001 |pmid=11259612 |doi= |url=}}</ref> | ||
* | * The mutated versions of [[Prokineticin|PROK2]] and [[Prokineticin receptor 2|PROKR2]] could lead to decrease [[GnRH]] production and [[hypogonadism]]. Other disorders caused by [[Prokineticin|PROK2]] and [[Prokineticin receptor 2|PROKR2]] [[mutations]] include [[fibrous dysplasia]], [[sleep disorder]], severe [[obesity]], [[synkinesis]], and [[epilepsy]].<ref name="pmid18559922">{{cite journal |vauthors=Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Raivio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley WF, Zhou QY, Pitteloud N |title=Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum |journal=J. Clin. Endocrinol. Metab. |volume=93 |issue=9 |pages=3551–9 |year=2008 |pmid=18559922 |pmc=2567850 |doi=10.1210/jc.2007-2654 |url=}}</ref> | ||
=== Tachykinin 3 and tachykinin 3 receptor (TAC3 and TACR3) === | === Tachykinin 3 and tachykinin 3 receptor (TAC3 and TACR3) === | ||
* The [[Tachykinin|TAC3]] and [[Tachykinin receptor 3|TACR3]] [[genes]], also called [[Neurokinin B|neurokinin B (NKB)]] and [[neurokinin]] 3 receptor (NK3R), with [[OMIM]] numbers of 162330 and 152332, are on [[chromosomes]] 12q13–q21 and 4q25, respectively.<ref name="pmid19079066">{{cite journal |vauthors=Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK |title=TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction |journal=Nat. Genet. |volume=41 |issue=3 |pages=354–358 |year=2009 |pmid=19079066 |pmc=4312696 |doi=10.1038/ng.306 |url=}}</ref> | * The [[Tachykinin|TAC3]] and [[Tachykinin receptor 3|TACR3]] [[genes]], also called [[Neurokinin B|neurokinin B (NKB)]] and [[neurokinin]] 3 receptor (NK3R), with [[OMIM]] numbers of 162330 and 152332, are on [[chromosomes]] 12q13–q21 and 4q25, respectively.<ref name="pmid19079066">{{cite journal |vauthors=Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK |title=TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction |journal=Nat. Genet. |volume=41 |issue=3 |pages=354–358 |year=2009 |pmid=19079066 |pmc=4312696 |doi=10.1038/ng.306 |url=}}</ref> | ||
* | * It is postulated that the normal function of [[Tachykinin|TAC3]]/[[Tachykinin receptor 3|TACR3]] system is necessary for an intact HPG axis and its development during [[puberty]]. On the other hand, [[Tachykinin|TAC3]]/[[Tachykinin receptor 3|TACR3]] system disturbance may cause [[micropenis]] and [[cryptorchidism]] in males, showing the major role of [[Tachykinin|TAC3]]/[[Tachykinin receptor 3|TACR3]] in fetal [[gonadotropins]] secretion.<ref name="pmid15212980">{{cite journal |vauthors=Pinto FM, Almeida TA, Hernandez M, Devillier P, Advenier C, Candenas ML |title=mRNA expression of tachykinins and tachykinin receptors in different human tissues |journal=Eur. J. Pharmacol. |volume=494 |issue=2-3 |pages=233–9 |year=2004 |pmid=15212980 |doi=10.1016/j.ejphar.2004.05.016 |url=}}</ref> | ||
* [[Tachykinin receptor 3|TACR3]] encoded protein (NK3R) is [[G protein-coupled receptor|GPCR]], initially produced in [[central nervous system]]. The major mechanism, through which the mutated [[gene]] may lead to [[neuroendocrine]] disturbance and delayed [[puberty]], is not completely | * [[Tachykinin receptor 3|TACR3]] encoded protein (NK3R) is [[G protein-coupled receptor|GPCR]], initially produced in [[central nervous system]]. The major mechanism, through which the mutated [[gene]] may lead to [[neuroendocrine]] disturbance and delayed [[puberty]], is not yet discovered completely.<ref name="pmid19719764">{{cite journal |vauthors=Semple RK, Topaloglu AK |title=The recent genetics of hypogonadotrophic hypogonadism - novel insights and new questions |journal=Clin. Endocrinol. (Oxf) |volume=72 |issue=4 |pages=427–35 |year=2010 |pmid=19719764 |doi=10.1111/j.1365-2265.2009.03687.x |url=}}</ref> | ||
* [[Tachykinin|TAC3]] encoded protein (NKB) is produced in [[arcuate nucleus]] of [[hypothalamus]] and play an important role in [[GnRH]] secretion. | * [[Tachykinin|TAC3]] encoded protein (NKB) is produced in [[arcuate nucleus]] of [[hypothalamus]] and play an important role in [[GnRH]] secretion. [[Kisspeptin]] is also produced and secreted in [[arcuate nucleus]], whereas, both of them are inhibited by [[estrogen]]. It may be considered that [[kisspeptin]] and [[Neurokinin B|NKB]] have identical roles in diverting [[negative feedback]] from [[sex hormones]] to [[GnRH]]. Their mutation has been shown to be related with [[hypogonadism]]. | ||
=== Gonadotropin releasing hormone and its receptor (GnRH1 and GnRHR) === | === Gonadotropin releasing hormone and its receptor (GnRH1 and GnRHR) === | ||
* The [[Gonadotropin-releasing hormone|GnRH1]] and [[GnRHR]] [[genes]] with [[OMIM]] numbers of 152760 and 138850 are on [[chromosomes]] 8p21–8p11.2 and 4q21.2, respectively.<ref name="pmid19535795">{{cite journal |vauthors=Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J |title=Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation |journal=N. Engl. J. Med. |volume=360 |issue=26 |pages=2742–8 |year=2009 |pmid=19535795 |doi=10.1056/NEJMoa0900136 |url=}}</ref> | * The [[Gonadotropin-releasing hormone|GnRH1]] and [[GnRHR]] [[genes]] with [[OMIM]] numbers of 152760 and 138850 are on [[chromosomes]] 8p21–8p11.2 and 4q21.2, respectively.<ref name="pmid19535795">{{cite journal |vauthors=Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J |title=Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation |journal=N. Engl. J. Med. |volume=360 |issue=26 |pages=2742–8 |year=2009 |pmid=19535795 |doi=10.1056/NEJMoa0900136 |url=}}</ref> | ||
* In HPG axis, [[GnRH]] is one of the most effective elements; therefore, | * In HPG axis, [[GnRH]] is one of the most effective elements; therefore, a defect could directly influence the axis and slow down the progress. | ||
* The GnRHR [[gene]] is also responsible for [[gonadal]] normal functions | * The GnRHR [[gene]] is also responsible for [[gonadal]] normal functions and its mutation could lead to [[hypogonadism]] and delayed [[puberty]]. The [[mutation]] in GnRHR has also been associated with conditions such as [[atrophic]] [[gonads]] along with low [[LH]]/[[FSH]] and [[sex hormones]], sexual [[puberty]] disturbance, inability to [[Conceive a child|conceive]], and failure to impact from exogenous [[GnRH]].<ref name="pmid20068010">{{cite journal |vauthors=Wu S, Wilson MD, Busby ER, Isaac ER, Sherwood NM |title=Disruption of the single copy gonadotropin-releasing hormone receptor in mice by gene trap: severe reduction of reproductive organs and functions in developing and adult mice |journal=Endocrinology |volume=151 |issue=3 |pages=1142–52 |year=2010 |pmid=20068010 |doi=10.1210/en.2009-0598 |url=}}</ref> | ||
* | * [[Gonadotropin-releasing hormone|GnRH1]] and [[GnRHR]] [[genes]] have variable expression and cause a spectrum of symptoms, from fertile eunuch syndrome and partial idiopathic [[hypogonadotropic hypogonadism]] to complete [[GnRH]] resistance (i.e., characterized by [[cryptorchidism]]), [[microphallus]], very low [[LH]]/[[FSH]], and delayed [[puberty]].<ref name="pmid12536356">{{cite journal |vauthors=Silveira LF, MacColl GS, Bouloux PM |title=Hypogonadotropic hypogonadism |journal=Semin. Reprod. Med. |volume=20 |issue=4 |pages=327–38 |year=2002 |pmid=12536356 |doi=10.1055/s-2002-36707 |url=}}</ref> | ||
* The other disorders that have found to be related to [[GnRH]] mutation are | * The other disorders that have been found to be related to [[GnRH]] mutation are [[tooth]] [[maturation]] and biomineralization.<ref name="pmid17948256">{{cite journal |vauthors=Tiong J, Locastro T, Wray S |title=Gonadotropin-releasing hormone-1 (GnRH-1) is involved in tooth maturation and biomineralization |journal=Dev. Dyn. |volume=236 |issue=11 |pages=2980–92 |year=2007 |pmid=17948256 |doi=10.1002/dvdy.21332 |url=}}</ref> | ||
=== Chromodomain helicase DNA-binding protein 7 (CHD7) === | === Chromodomain helicase DNA-binding protein 7 (CHD7) === | ||
* The [[CHD7]] gene, also called as KAL5, with [[OMIM]] number of 608892 is on [[chromosome]] 8q12.1. | * The [[CHD7]] gene, also called as KAL5, with [[OMIM]] number of 608892 is on [[chromosome]] 8q12.1. | ||
* | * [[CHD7]] gene [[mutation]] results in [[autosomal dominant]] [[CHARGE syndrome]], which is a combination of [[hypogonadism]] and [[Kallman syndrome|Kallmann syndrome]], and includes:<ref name="pmid188349672">{{cite journal |vauthors=Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC |title=Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome |journal=Am. J. Hum. Genet. |volume=83 |issue=4 |pages=511–9 |year=2008 |pmid=18834967 |pmc=2561938 |doi=10.1016/j.ajhg.2008.09.005 |url=}}</ref> | ||
** [[Coloboma|'''C''' | ** [[Coloboma|'''C'''oloboma]] | ||
** [[Heart|'''H'''eart]] anomalies | ** [[Heart|'''H'''eart]] anomalies | ||
** [[Choanal atresia|Choanal '''A'''tresia]] | ** [[Choanal atresia|Choanal '''A'''tresia]] | ||
| Line 410: | Line 417: | ||
** [[Genital|'''G'''enital]] anomalies | ** [[Genital|'''G'''enital]] anomalies | ||
** [[Ear|'''E'''ar]] anomalies | ** [[Ear|'''E'''ar]] anomalies | ||
* | * Screening for [[CHD7]] [[gene]] mutation may be done in patients with [[hypogonadism]] or [[Kallman syndrome|Kallmann syndrome]] with specific features such as [[semicircular canals|semicircular canal]] [[hypoplasia]] or [[aplasia]], [[dysmorphic]] ears, and [[deafness|deafness.]] | ||
=== Nasal embryonic LH-releasing hormone factor (NELF) === | === Nasal embryonic LH-releasing hormone factor (NELF) === | ||
* The NELF [[gene]] with [[OMIM]] number of 608137 is on [[chromosome]] ''9q34.3; | * The NELF [[gene]] with [[OMIM]] number of 608137 is on [[chromosome]] ''9q34.3''; present mostly in [[nervous tissues]] specifically during [[fetal development]] and may be found in [[olfactory bulb]] and [[pituitary]] [[LH]] releasing cells. | ||
* | * The most common function is in [[olfactory]] axons and [[GnRH]] [[neurons]], before and during [[neuron]] migration in the developmental process.<ref name="pmid108987962">{{cite journal |vauthors=Kramer PR, Wray S |title=Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons |journal=Genes Dev. |volume=14 |issue=14 |pages=1824–34 |year=2000 |pmid=10898796 |pmc=316793 |doi= |url=}}</ref> | ||
=== Early B-cell factor 2 (EBF2) === | === Early B-cell factor 2 (EBF2) === | ||
* The EBF2 [[gene]] with [[OMIM]] number of 609934 is on [[chromosome]] ''8p21.2; mostly expressed in mice [[osteoblasts]] and [[osteoclast]] cells. | * The EBF2 [[gene]] with [[OMIM]] number of 609934 is on [[chromosome]] ''8p21.2''; mostly expressed in mice [[osteoblasts]] and [[osteoclast]] cells.<ref name="pmid12466206">{{cite journal |vauthors=Corradi A, Croci L, Broccoli V, Zecchini S, Previtali S, Wurst W, Amadio S, Maggi R, Quattrini A, Consalez GG |title=Hypogonadotropic hypogonadism and peripheral neuropathy in Ebf2-null mice |journal=Development |volume=130 |issue=2 |pages=401–10 |year=2003 |pmid=12466206 |doi= |url=}}</ref> | ||
* | * EBF2 [[gene]] plays an effective role in HPG axis. Mutation in EBF2 [[gene]] can result in disruption of HPG axis, leading to secondary [[hypogonadism]].<ref name="pmid16423815">{{cite journal |vauthors=Trarbach EB, Baptista MT, Garmes HM, Hackel C |title=Molecular analysis of KAL-1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients |journal=J. Endocrinol. |volume=187 |issue=3 |pages=361–8 |year=2005 |pmid=16423815 |doi=10.1677/joe.1.06103 |url=}}</ref> | ||
=== DSS-AHC on the X-chromosome 1 (DAX1) === | === DSS-AHC on the X-chromosome 1 (DAX1) === | ||
* The [[DAX1]] [[gene]], also called [[nuclear receptor]] 0B (NR0B), with [[OMIM]] number of 300473 is on [[chromosome]] ''Xp21.2, | * The [[DAX1]] [[gene]], also called [[nuclear receptor]] 0B (NR0B), with [[OMIM]] number of 300473 is on [[chromosome]] ''Xp21.2'', and expressed in all members of HPG axis ([[hypothalamus]], [[pituitary]], and [[gonads]]).<ref name="pmid8593542">{{cite journal |vauthors=Guo W, Burris TP, McCabe ER |title=Expression of DAX-1, the gene responsible for X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism, in the hypothalamic-pituitary-adrenal/gonadal axis |journal=Biochem. Mol. Med. |volume=56 |issue=1 |pages=8–13 |year=1995 |pmid=8593542 |doi= |url=}}</ref> | ||
* During the [[spermatogenesis]] and [[steroidogenesis]], | * During the [[spermatogenesis]] and [[steroidogenesis]], both [[Sertoli cell|sertoli]] and [[leydig cells]] have increased expression of [[DAX1]] gene. It is assumed that during [[puberty]], the peak expression of [[DAX1]] is observed.<ref name="pmid16834661">{{cite journal |vauthors=Kojima Y, Sasaki S, Hayashi Y, Umemoto Y, Morohashi K, Kohri K |title=Role of transcription factors Ad4bp/SF-1 and DAX-1 in steroidogenesis and spermatogenesis in human testicular development and idiopathic azoospermia |journal=Int. J. Urol. |volume=13 |issue=6 |pages=785–93 |year=2006 |pmid=16834661 |doi=10.1111/j.1442-2042.2006.01403.x |url=}}</ref> | ||
* Other [[disease]] that can be caused by [[DAX1]] mutation is congenital [[Adrenal cortex insufficiency|adrenal cortex hypoplasia]].<ref name="pmid7990953">{{cite journal |vauthors=Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER |title=An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita |journal=Nature |volume=372 |issue=6507 |pages=635–41 |year=1994 |pmid=7990953 |doi=10.1038/372635a0 |url=}}</ref> | * Other [[disease]] that can be caused by [[DAX1]] mutation is congenital [[Adrenal cortex insufficiency|adrenal cortex hypoplasia]].<ref name="pmid7990953">{{cite journal |vauthors=Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER |title=An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita |journal=Nature |volume=372 |issue=6507 |pages=635–41 |year=1994 |pmid=7990953 |doi=10.1038/372635a0 |url=}}</ref> | ||
=== Steroidogenic factor 1 (SF1) === | === Steroidogenic factor 1 (SF1) === | ||
* The [[SF1 (gene)|SF1]] [[gene]], also called [[nuclear receptor]] 5A1 (NR5A1), with [[OMIM]] number of 184757 is on [[chromosome]] | * The [[SF1 (gene)|SF1]] [[gene]], also called [[nuclear receptor]] 5A1 (NR5A1), with [[OMIM]] number of 184757 is on [[chromosome]] 9q33.3, has some role in [[reproduction]], [[steroidogenesis]], and [[sexual differentiation]]. | ||
* It is mainly expressed in [[Sertoli cell|sertoli]] and [[leydig cells]], plays an important role in [[steroidogenesis]] and [[spermatogenesis]]. The [[SF1]] is believed to | * It is mainly expressed in [[Sertoli cell|sertoli]] and [[leydig cells]], and plays an important role in [[steroidogenesis]] and [[spermatogenesis]]. The [[SF1]] is believed to have an increase in expression from [[childhood]] until [[adolescence]], and is dominantly expressed by [[leydig cells]] in [[puberty]].<ref name="pmid16834661" /> | ||
* | * Other diseases that may be caused by [[SF1 (gene)|SF1]] mutation include male [[pseudohermaphroditism]], [[Denys-Drash syndrome]], and also [[hypospadias]].<ref name="pmid9590178">{{cite journal |vauthors=Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan JN, Hammer GD, Ingraham HA |title=Wilms' tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression |journal=Cell |volume=93 |issue=3 |pages=445–54 |year=1998 |pmid=9590178 |doi= |url=}}</ref> | ||
=== Homeobox gene 1 (HESX1) === | === Homeobox gene 1 (HESX1) === | ||
* The [[HESX1]] [[gene]], also called [[Rathke pouch]] [[Homeobox gene|homeobox]] (RPX), with [[OMIM]] number of 601802 is on [[chromosome]] | * The [[HESX1]] [[gene]], also called [[Rathke pouch]] [[Homeobox gene|homeobox]] (RPX), with [[OMIM]] number of 601802 is on [[chromosome]] 3p14.3, initially expressed during [[embryogenesis]] and help the formation of [[Rathke pouch]] and [[anterior pituitary]]''.''<ref name="pmid9620767">{{cite journal |vauthors=Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Mårtensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC |title=Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse |journal=Nat. Genet. |volume=19 |issue=2 |pages=125–33 |year=1998 |pmid=9620767 |doi=10.1038/477 |url=}}</ref> | ||
* | * [[HESX1]] gene has an important role in [[pituitary]] development and midfacial differentiation. [[Mutation]] may lead to [[pituitary]] [[hypoplasia]] and decreased level of all [[anterior pituitary]] [[hormones]].<ref name="pmid11136712">{{cite journal |vauthors=Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS |title=Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia |journal=Hum. Mol. Genet. |volume=10 |issue=1 |pages=39–45 |year=2001 |pmid=11136712 |doi= |url=}}</ref> | ||
* Other disorders resulting from [[HESX1]] [[mutation]] | * Other disorders resulting from [[HESX1]] [[mutation]] <nowiki/>include septo optic dysplasia, reduced [[prosencephalon]], [[anophthalmia]], [[microphthalmia]], defective [[olfactory]] development, [[Rathke pouch]] bifurcations, and abnormalities in the [[corpus callosum]], [[hippocampus]], and [[septum pellucidum]].<ref name="pmid9620767" /> | ||
=== LIM homeobox gene 3 (LHX3) === | === LIM homeobox gene 3 (LHX3) === | ||
* The [[LHX3|LHX3 gene]], also called LIM3, with [[OMIM]] number of 600577 is on [[chromosome]] | * The [[LHX3|LHX3 gene]], also called LIM3, with [[OMIM]] number of 600577 is on [[chromosome]] 9q34.3, mainly expressed in developing anterior [[pituitary gland]].<ref name="pmid18407919">{{cite journal |vauthors=Rajab A, Kelberman D, de Castro SC, Biebermann H, Shaikh H, Pearce K, Hall CM, Shaikh G, Gerrelli D, Grueters A, Krude H, Dattani MT |title=Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss |journal=Hum. Mol. Genet. |volume=17 |issue=14 |pages=2150–9 |year=2008 |pmid=18407919 |doi=10.1093/hmg/ddn114 |url=}}</ref> | ||
* | * [[LHX3|LHX3 gene]] function is important in development of [[pituitary gland]] and pituitary [[hormone|hormones]] secretion. Mutation in the [[LHX3|LHX3 gene]] may result in combined pituitary hormone deficiency (CPHD).<ref name="pmid10835633">{{cite journal |vauthors=Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers Av, Goossens M, Grüters A, Amselem S |title=Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency |journal=Nat. Genet. |volume=25 |issue=2 |pages=182–6 |year=2000 |pmid=10835633 |doi=10.1038/76041 |url=}}</ref> | ||
* | * [[LHX3|LHX3 gene]] [[mutation]] may also result in neonatal [[hypoglycemia]], short neck with limited rotation, mild [[Sensorineural hearing loss|sensorineural hearing loss,]] skin laxity, and skeletal abnormalities.<ref name="pmid18407919" /> | ||
=== PROP paired-like homeobox 1 (PROP1) === | === PROP paired-like homeobox 1 (PROP1) === | ||
* The [[PROP1|PROP1 gene]] with [[OMIM]] number of 601538 is on [[chromosome]] | * The [[PROP1|PROP1 gene]] with [[OMIM]] number of 601538 is on [[chromosome]] 5q35.3, with a role in developing anterior [[pituitary gland]] and associated cells such as [[gonadotrophs]], [[thyrotrophs]], [[somatotrophs]], and [[Lactotrophs|lactotrophs.]]<ref name="pmid9824293">{{cite journal |vauthors=Duquesnoy P, Roy A, Dastot F, Ghali I, Teinturier C, Netchine I, Cacheux V, Hafez M, Salah N, Chaussain JL, Goossens M, Bougnères P, Amselem S |title=Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency |journal=FEBS Lett. |volume=437 |issue=3 |pages=216–20 |year=1998 |pmid=9824293 |doi= |url=}}</ref> | ||
* | * Mutated [[PROP1|PROP1 gene]] can lead to deficiency of [[LH]], [[FSH]], [[GH]], [[TSH]], and [[prolactin]]. Decreased level of [[LH]] and [[FSH]] may also delay or inhibit the onset of [[puberty]].<ref name="pmid9462743">{{cite journal |vauthors=Wu W, Cogan JD, Pfäffle RW, Dasen JS, Frisch H, O'Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA, Rosenfeld MG |title=Mutations in PROP1 cause familial combined pituitary hormone deficiency |journal=Nat. Genet. |volume=18 |issue=2 |pages=147–9 |year=1998 |pmid=9462743 |doi=10.1038/ng0298-147 |url=}}</ref> | ||
* | * [[Pituitary]] [[Hormone|hormones]] have a vital role in regulating other endocrine organs via [[TRH]], [[Adrenocorticotropic hormone|ACTH]], [[FSH]] or [[LH]] and a mutation in [[PROP1|PROP1 gene]] can lead to [[thyroid]] dysfunctions, [[growth retardation]], and [[libido]]/[[lactation]] problems. | ||
=== Leptin and leptin receptor (LEP and LEPR) === | === Leptin and leptin receptor (LEP and LEPR) === | ||
* The [[LEP]] and [[LEPR|LEPR genes]], also called OB and OBR, with [[OMIM]] numbers of 164160 and 601007 are on [[chromosome]]<nowiki/>s | * The [[LEP]] and [[LEPR|LEPR genes]], also called OB and OBR, with [[OMIM]] numbers of 164160 and 601007 are on [[chromosome]]<nowiki/>s 7q32.1 and 1p31.3, respectively; with a major role in modulation of [[body weight]][[Lactotrophs|.]] | ||
* These [[genes]] are believed to carry the message of | * These [[genes]] are believed to carry the message of onset of [[puberty]]. Recent studies have shown that [[recombinant]] [[leptin]] injection in female mice may result in [[puberty]] and cure their [[maturation]] ([[secondary sexual characteristics]]) problems.<ref name="pmid8589726">{{cite journal |vauthors=Chehab FF, Lim ME, Lu R |title=Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin |journal=Nat. Genet. |volume=12 |issue=3 |pages=318–20 |year=1996 |pmid=8589726 |doi=10.1038/ng0396-318 |url=}}</ref> | ||
* It | * It has been observed that [[leptin]] levels increase by 50% just before the onset of [[puberty]] and during [[puberty]].<ref name="pmid9100574">{{cite journal |vauthors=Mantzoros CS, Flier JS, Rogol AD |title=A longitudinal assessment of hormonal and physical alterations during normal puberty in boys. V. Rising leptin levels may signal the onset of puberty |journal=J. Clin. Endocrinol. Metab. |volume=82 |issue=4 |pages=1066–70 |year=1997 |pmid=9100574 |doi=10.1210/jcem.82.4.3878 |url=}}</ref> | ||
* [[Mutation]] in | * [[Mutation]] in [[LEP]] and [[LEPR|LEPR genes]] may result in dysfunctional [[hematopoiesis]], [[angiogenesis]], [[wound healing]], and the [[immune]] or [[inflammatory response]]. | ||
=== Proprotein | === Proprotein convertase 1 (PC1) === | ||
* The PC1 [[gene]], also | * The PC1 [[gene]], also known as [[neuroendocrine]] convertase 1 (NEC1), with [[OMIM]] number of 162150 is on [[chromosome]] 5q15, and regulates [[neuroendocrine]] pathway. | ||
* PC1 gene has | * PC1 gene has a dominant role in [[proopiomelanocortin]] (POMC) cleavage. PC1 gene also has a role in processing [[proinsulin]] and [[proglucagon]] in [[pancreas]].<ref name="pmid7797529">{{cite journal |vauthors=Jansen E, Ayoubi TA, Meulemans SM, Van de Ven WJ |title=Neuroendocrine-specific expression of the human prohormone convertase 1 gene. Hormonal regulation of transcription through distinct cAMP response elements |journal=J. Biol. Chem. |volume=270 |issue=25 |pages=15391–7 |year=1995 |pmid=7797529 |doi= |url=}}</ref> | ||
* | * Recent studies have shown that PC1 [[gene]] [[mutation]] and [[hypogonadotropic hypogonadism]] may result in extreme childhood [[obesity]], abnormal glucose [[homeostasis]], [[hypocortisolism]], elevated plasma [[proinsulin]], and [[POMC]] concentrations.<ref name="pmid9207799">{{cite journal |vauthors=Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S |title=Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene |journal=Nat. Genet. |volume=16 |issue=3 |pages=303–6 |year=1997 |pmid=9207799 |doi=10.1038/ng0797-303 |url=}}</ref> | ||
=== Makorin RING-finger protein 3 (MKRN3) === | === Makorin RING-finger protein 3 (MKRN3) === | ||
* Newly discovered MKRN3 gene has a role in [[ubiquitination]] and [[cell signaling]]. The gene family [[proteins]] are majorly expressed in fetal [[brain]] during development, especially in [[arcuate nucleus]]. | * Newly discovered MKRN3 gene has a role in [[ubiquitination]] and [[cell signaling]]. The gene family [[proteins]] are majorly expressed in fetal [[brain]] during development, especially in [[arcuate nucleus]]. | ||
* | * The process of gene amplification is on its peak after [[birth]], which gradually declines with time, and finally rises again with onset of [[puberty]]. Thus MKRN3 gene is believed to be one of the factors in onset of [[puberty]], along with [[Kisspeptin|kisspeptins]] and [[neurokinin B]].<ref name="Hughes2013">{{cite journal|last1=Hughes|first1=Ieuan A.|title=Releasing the Brake on Puberty|journal=New England Journal of Medicine|volume=368|issue=26|year=2013|pages=2513–2515|issn=0028-4793|doi=10.1056/NEJMe1306743}}</ref> | ||
=== Estrogen receptor α (ESR1) === | === Estrogen receptor α (ESR1) === | ||
* [[Estrogen receptor]] [[mutations]] are very rare, reported | * [[Estrogen receptor]] [[mutations]] are very rare, and were reported in a case report of delayed [[puberty]].<ref name="QuaynorStradtman2013">{{cite journal|last1=Quaynor|first1=Samuel D.|last2=Stradtman|first2=Earl W.|last3=Kim|first3=Hyung-Goo|last4=Shen|first4=Yiping|last5=Chorich|first5=Lynn P.|last6=Schreihofer|first6=Derek A.|last7=Layman|first7=Lawrence C.|title=Delayed Puberty and Estrogen Resistance in a Woman with Estrogen Receptor α Variant|journal=New England Journal of Medicine|volume=369|issue=2|year=2013|pages=164–171|issn=0028-4793|doi=10.1056/NEJMoa1303611}}</ref> | ||
* [[Estradiol]] | * [[Estradiol]] promotes [[breast]] maturation and provides [[negative feedback]] to [[hypothalamus]] and [[pituitary]], by means of [[Estrogen receptor alpha|estrogen receptor α]] (encoded by ESR1 [[gene]]).<ref name="pmid18635656">{{cite journal |vauthors=Christian CA, Glidewell-Kenney C, Jameson JL, Moenter SM |title=Classical estrogen receptor alpha signaling mediates negative and positive feedback on gonadotropin-releasing hormone neuron firing |journal=Endocrinology |volume=149 |issue=11 |pages=5328–34 |year=2008 |pmid=18635656 |pmc=2584581 |doi=10.1210/en.2008-0520 |url=}}</ref> | ||
* Female mice with mutated ESR1 [[gene]] | * Female mice with mutated ESR1 [[gene]] have [[hypoplastic]] uterus with [[hemorrhagic|hemorrhage]], multicystic [[ovary]] without [[corpus luteum]]; which is make them [[infertile]].<ref name="pmid8248223">{{cite journal |vauthors=Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O |title=Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=90 |issue=23 |pages=11162–6 |year=1993 |pmid=8248223 |pmc=47942 |doi= |url=}}</ref> | ||
==Associated Conditions== | ==Associated Conditions== | ||
| Line 474: | Line 480: | ||
{{Family tree | | | C01 | | | | C02 | | | | C03 | | C01= '''''Primary [[amenorrhea]]'''''| C02= '''''Secondary [[amenorrhea]]'''''| C03= '''''Functional [[amenorrhea]]'''''}} | {{Family tree | | | C01 | | | | C02 | | | | C03 | | C01= '''''Primary [[amenorrhea]]'''''| C02= '''''Secondary [[amenorrhea]]'''''| C03= '''''Functional [[amenorrhea]]'''''}} | ||
{{Family tree | | | |!| | | | | |!| | | | | |!| | }} | {{Family tree | | | |!| | | | | |!| | | | | |!| | }} | ||
{{Family tree |boxstyle=text-align: left; | | | C01 | | | | C02 | | | | C03 | | C01=• [[Kallmann syndrome]]<br> • [[Turner syndrome]]<br> • [[Noonan syndrome]]<br> • [[Gonadal dysgenesis]]<br> • [[Chemotherapy]]/[[Radiation therapy]]<br> • [[Coxsackie]]<br> • [[Galactosemia]]<br> • Autoimmune oophiritis<br> • [[Adenylosuccinate lyase deficiency|Lyase deficiency]]<br> • [[Congenital lipoid adrenal hyperplasia]]<br> • [[Androgen insensitivity]]<br> • Congenital [[hypopituitarism]]<br> • [[Bardet-Biedl syndrome]]<br> • [[CHARGE syndrome]]<br> • [[Gaucher disease]]<br> • [[Septo-optic dysplasia]]<br>• [[Cystic Fibrosis]]<br> • [[Thalassemia]]| C02= • [[Astrocytoma]]<br> • [[Germinoma]]<br> • [[Glioma]]<br> • [[Craniopharyngioma]]<br> • [[Prolactinoma]]<br> • [[Langerhans cell histiocytosis]]<br> • [[Rathke pouch]] cyst<br> • Isolated hypogonadotropic [[hypogonadism]] <br> • [[Hypothalamic-pituitary-gonadal axis|HPO axis]] development disturbance<br> • Post [[central nervous system]] [[Infection]]<br> • [[Chemotherapy]]/[[Radiation therapy]]<br> • [[Trauma]]<br> • [[Asthma]] <br> • [[Inflammatory bowel disease]] <br> • [[Celiac disease]] <br> • [[Juvenile rheumatoid arthritis]]<br> • [[Sickle cell disease]]<br> • [[Hemosiderosis]]<br> • [[Chronic renal disease]]<br> • [[AIDS]]<br> • [[Diabetes mellitus]] <br> • [[Hypothyroidism]]<br> • [[Hyperprolactinemia]]<br> • [[Growth hormone deficiency]] <br> • [[Cushing syndrome]]|C03=• [[Stress]]<br> • Excessive [[exercise]]<br> • [[Malnutrition]]<br> • [[Obesity]] <br> • [[Anorexia nervosa]]<br> • [[Bulimia]] }} | {{Family tree |boxstyle=text-align: left; | | | C01 | | | | C02 | | | | C03 | | C01=• [[Kallmann syndrome]]<br> • [[Turner syndrome]]<br> • [[Noonan syndrome]]<br> • [[Gonadal dysgenesis]]<br> • [[Chemotherapy]]/[[Radiation therapy]]<br> • [[Coxsackie]]<br> • [[Galactosemia]]<br> • [[Autoimmune]] [[oophiritis]]<br> • [[Adenylosuccinate lyase deficiency|Lyase deficiency]]<br> • [[Congenital lipoid adrenal hyperplasia]]<br> • [[Androgen insensitivity]]<br> • [[Congenital]] [[hypopituitarism]]<br> • [[Bardet-Biedl syndrome]]<br> • [[CHARGE syndrome]]<br> • [[Gaucher disease]]<br> • [[Septo-optic dysplasia]]<br>• [[Cystic Fibrosis]]<br> • [[Thalassemia]]| C02= • [[Astrocytoma]]<br> • [[Germinoma]]<br> • [[Glioma]]<br> • [[Craniopharyngioma]]<br> • [[Prolactinoma]]<br> • [[Langerhans cell histiocytosis]]<br> • [[Rathke pouch]] cyst<br> • Isolated hypogonadotropic [[hypogonadism]] <br> • [[Hypothalamic-pituitary-gonadal axis|HPO axis]] development disturbance<br> • Post [[central nervous system]] [[Infection]]<br> • [[Chemotherapy]]/[[Radiation therapy]]<br> • [[Trauma]]<br> • [[Asthma]] <br> • [[Inflammatory bowel disease]] <br> • [[Celiac disease]] <br> • [[Juvenile rheumatoid arthritis]]<br> • [[Sickle cell disease]]<br> • [[Hemosiderosis]]<br> • [[Chronic renal disease]]<br> • [[AIDS]]<br> • [[Diabetes mellitus]] <br> • [[Hypothyroidism]]<br> • [[Hyperprolactinemia]]<br> • [[Growth hormone deficiency]] <br> • [[Cushing syndrome]]|C03=• [[Stress]]<br> • Excessive [[exercise]]<br> • [[Malnutrition]]<br> • [[Obesity]] <br> • [[Anorexia nervosa]]<br> • [[Bulimia]] }} | ||
{{Family tree/end}} | {{Family tree/end}} | ||
| Line 482: | Line 488: | ||
*On [[gross pathology]], normal [[endometrium]] in proliferative or [[Luteal phase|luteal phases]] are characteristic findings of amenorrhea. | *On [[gross pathology]], normal [[endometrium]] in proliferative or [[Luteal phase|luteal phases]] are characteristic findings of amenorrhea. | ||
*In | *In cases of amenorrhea which are secondary to other causes, the related [[gross pathology]] would be seen. | ||
*[[Craniopharyngioma]] [[gross pathology]] | *[[Craniopharyngioma]] on [[gross pathology]] presents as cystic mass filled with motor oil-like fluid.<ref name="pmid21584897">{{cite journal |vauthors=Fernandez-Miranda JC, Gardner PA, Snyderman CH, Devaney KO, Strojan P, Suárez C, Genden EM, Rinaldo A, Ferlito A |title=Craniopharyngioma: a pathologic, clinical, and surgical review |journal=Head Neck |volume=34 |issue=7 |pages=1036–44 |year=2012 |pmid=21584897 |doi=10.1002/hed.21771 |url=}}</ref> <br> <br><br><br><br><br> <br><br><br><br><br> <br><br><br> <br> | ||
==Microscopic Pathology== | ==Microscopic Pathology== | ||
*On [[microscopic]] [[histopathological]] analysis, | *On [[microscopic]] [[histopathological]] analysis, [[craniopharyngioma]] as a cause of amenorrhea will have the following features: | ||
*On [[microscopic]] [[histopathological]] analysis, | **Trabecular [[squamous epithelium]] surrounded by palisaded [[columnar epithelium]] | ||
**Small-to-medium sized cells with moderate amount of [[basophilic]] [[cytoplasm]] | |||
**Bland [[nuclei]] | |||
**[[Calcification|Calcifications]] | |||
*On [[microscopic]] [[histopathological]] analysis, [[pituitary adenoma]] as a cause of amenorrhea will have the following features: | |||
**Loss of [[fibrous]] [[stroma]] | |||
**Nested cells of normal [[anterior pituitary]] (based on the type of [[adenoma]]) | |||
<gallery align="left"> | <gallery align="left"> | ||
| Line 504: | Line 516: | ||
{{WH}} | {{WH}} | ||
{{WS}} | {{WS}} | ||
[[Category:Medicine]] | |||
[[Category:Endocrinology]] | |||
[[Category:Up-To-Date]] | |||
[[Category:Gynecology]] | |||
[[Category:Obstetrics]] | |||
Latest revision as of 20:22, 29 July 2020
|
Amenorrhea Microchapters |
|
Patient Information |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Amenorrhea pathophysiology On the Web |
|
American Roentgen Ray Society Images of Amenorrhea pathophysiology |
|
Risk calculators and risk factors for Amenorrhea pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Eiman Ghaffarpasand, M.D. [2]
Overview
Amenorrhea is defined as absence of menstrual cycle. The causes of amenorrhea include hypothalamic, pituitary, thyroid, adrenal, ovarian, uterine, and vaginal. About 25 different genes are involved in the pathogenesis of amenorrhea including 3 different groups of Kallmann syndrome related genes, hypothalamus-pituitary-gonadal (HPG) axis related genes, and obesity related genes. On gross pathology, normal endometrium is the characteristic findings of amenorrhea. Patients of amenorrhea from Craniopharyngioma as have cystic mass filled with motor oil-like fluid on gross pathology. On microscopic histopathological analysis, craniopharyngioma presents as trabecular squamous epithelium surrounded by palisaded columnar epithelium, small-to-medium sized cells with moderate amount of basophilic cytoplasm, bland nuclei, and calcifications. On microscopic histopathological analysis, pituitary adenoma as a cause of amenorrhea presents as loss of fibrous stroma and nested cells of normal anterior pituitary (based on the type of adenoma).
Pathophysiology
Physiology of normal puberty
Menarche and Menstruation
- The mean age for onset of menstruation is 12.43 years in US. About 80% of females experience menarche between 11 and 13.75 years of age.[1]
- Almost all (98%) of females experience menarche by the age of 15.[2]
- Gonadotropin releasing hormone (GnRH) is the main factor in puberty onset.
- GnRH is secreted by the neurosecretory neurons of hypothalamus into the hypophysial portal system, from where it is transferred to anterior pituitary gland.
- GnRH stimulates production and secretion of follicle stimulating hormone (FSH) and luteinizing hormone (LH).
- During puberty the amplitude and frequency of GnRH pulses is significantly increased.
- GnRH secretion is regulated by certain neurotransmitters in brain, such as dopamine, endogenous opioids, norepinephrine, gamma amino butyric acid (GABA), and corticotropin releasing hormone (CRH). Alteration in level and function of these neurotransmitters may lead to specific types of amenorrhea. For example stress, exercise, and malnutrition affects CRH, β-endorphin, and dopamine, respectively.[3]
- After the onset of puberty, the negative feedback on GnRH is removed.
- Pulsatile secretion of GnRH induce LH and FSH production which finally lead to ovulation.
- In the absence of fertilisation, the ovarian follicle is turned into corpus luteum. Endometrium proliferates through estrogen release from corpus luteum. Withdrawal of progesterone from estrogen-mediated proliferated endometrium results in menstrual bleeding.
Hypothalamic-pituitary-ovarian (HPO) axis maturation
- After activation of the HPO axis during second trimester of pregnancy, the level of gonadotropins rises from mid to term pregnancy. After cessation of placental hormone feedback, FSH and LH increases again slightly to mild secondary peak.
- Before puberty, negative feedback from adrenal androgens keep the gonadotropins in low plasma level.[4]
- Right before puberty, the sensitivity of hypothalamus to negative feedback from adrenal androgens is decreased. This leads to increased production of GnRH from hypothalamus and make it possible for GnRH to be raised in magnitude and frequency to induce an increase in LH and FSH.[5]
- The complete maturation of HPO axis takes around 5-7 years from the onset of menstruation. Generally, during the first two years of menstruation, the cycles are mostly anovulatory due to immature HPO axis.
Pathogenesis
- Amenorrhea is defined as the absence of menstrual cycle.[3]
- Primary amenorrhea is the absence of menstrual cycle by 16 years of age, in the presence of normal growth and secondary sexual characteristics.
- Secondary amenorrhea reflects absence of menstrual cycle for at least 3 months in a woman with normal menstruation cycles in the past.
- The pathophysiology of amenorrhea is multifactorial and include hypothalamic, pituitary, thyroid, adrenal, ovarian, uterine, and vaginal causes.
Hypothalamic pathogenesis
- The most common cause of amenorrhea in adolescents are hypothalamic disorders and is known as hypothalamic amenorrhea.
- During the initial 2-3 years after the onset of menarche, the HPO axis is still under development. Immature HPO axis may lead to anovulatory cycles which can cause abnormalities in menstrual cycles.
- The most common cause of amenorrhea after 2-3 years of onset of puberty include eating disorders, excessive exercise, medications, and psychosocial stress.[6][7]
- Leptin plays an important role in energy consumption, body composition, food intake along with sexual maturation and reproductive improvement.
- It is assumed that leptin plays a role in the development of hypothalamic amenorrhea.
- Leptin receptors are in close relationship with hypothalamus and it is postulated that leptin regulates GnRH production and secretion.
- Patients with anorexia nervosa or excessive exercise have amenorrhea from down-regulation of leptin receptors, which can be elevated by refeeding.[8][9][10]
- Leptin level in cachexic patients will increase after gaining appropriate weight in patients without amenorrhea, but will remain low in patients with amenorrhea.[11][12]
- It has been observed that leptin levels in amenorrheic athletes are low as compared to non-athletes women or athletes with regular menses.[13][14]
- Administration of recombinant leptin for 3 months in women with conditions such as hypothalamic amenorrhea, excessive exercises or weight loss has been associated with an increased level of LH, FSH, and estradiol leading to ovulatory cycles.[15]
- Antipsychotic drugs and other medications that have an inhibitory effect on dopamine D2 receptor lead to an increased level of prolactin. Higher levels of prolactin suppress pulsatile GnRH secretion and block positive feedback of estradiol on hypothalamus, leading to disruption of HPO axis.[16]
- Stress and strenuous activities like other metabolic or cardiovascular responses are regulated through corticotropin releasing hormone (CRH), secreted by paraventricular nuclei of hypothalamus. CRH induce the release of β-endorphin, an endogenous opioid. Both CRH and β-endorphin suppress GnRH release. On the other hand, glucocorticoids suppress LH production from pituitary and also estrogen/progesterone from ovaries.[17]
- Kallmann syndrome, a genetic disorder caused by KAL gene mutation, has disturbance in migration of olfactory nerves along with GnRH neurons. Lack of GnRH leads to absence of secondary sexual characteristics and amenorrhea.[18]
Pituitary pathogenesis
- Prolactinoma is one of the most common anterior pituitary tumors. Prolactinoma leads to increased prolactin secretion and along with the tumor's mass effect may cause suppression of GnRH.
- The second most common tumor in the suprasellar region is craniopharyngioma. The tumor leads to LH and FSH disturbances, which may cause amenorrhea.[19]
Thyroid pathogenesis
- In hypothyroidism, the main mechanism that can lead to amenorrhea is the influence of thyrotropin releasing hormone (TRH) on lactotroph cells, increasing prolactin levels. Since the TRH is increased in hypothyroidism, it leads to functional hyperprolactinemia. The increase in prolactin may suppress the GnRH pulsatility and lead to amenorrhea.[20]
- In hyperthyroidism, the mechanism of amenorrhea is not clear. It is assumed that increased level of sex hormone binding globulins (SHBGs) in hyperthyroidism may lead to increased levels of androgens and estrogen. Conclusively, the LH surge become absent and amenorrhea happens.[21]
Adrenal pathogenesis
- Congenital adrenal hyperplasia (CAH) is a group of genetic enzyme deficiencies in adrenal gland. Most common is the defect in 21-hydroxylase enzyme, which leads to decrease in the level of aldosterone and cortisol. To overcome the hormone deficiencies, CRH production is increased by the hypothalamus. As described earlier, increased level of CRH may suppress GnRH and lead to amenorrhea.[22]
- Cushing syndrome has an increased level of cortisol, that can directly inhibit HPO axis and lead to amenorrhea.
Ovarian pathogenesis
- In polycystic ovary syndrome (PCOS) insulin resistance leads to increased androgen production (insulin reduces the SHBG circulating in plasma, causing increased testosterone). In ovaries, increased stimulation from GnRH leads to increased production of 17-hydroxy progesterone and cytochrome P450c17 which promotes androgens biosynthesis.[23][24] Finally, the pulsatility of GnRH will be disrupted leading to amenorrhea .[25]
- Primary ovarian insufficiency is multifactorial and leads to ovarian failure and decrease in estrogen which leads to amenorrhea. (In galactosemia, it is assumed that galactose and its metabolites are toxic to ovarian tissue.)[26]
Uterine pathogenesis
- The main pathogenesis of amenorrhea in androgen insensitivity syndrome is the absence of uterus. The patient is genotypically male, 46 XY; but with absent sexual characteristics due to lack of functional effect of androgen hormones on their receptors.
- One of the acquired conditions that can lead to amenorrhea is Asherman syndrome. The basis of Asherman syndrome is any condition that can alter the normal histology of endometrium, such as scarring from surgical procedure or adhesion from severe infection.
- Mayer-Rokitansky-Kuster-Hauser syndrome is complete agenesis of uterine, blind ended vagina. The lack of uterine and endometrium is the main pathogenesis of amenorrhea. The main reason of uterine agenesis is overactivation of anti-mullerian hormone in embryogenesis period.[27] Cervical agenesis also follow the similar process.
- Imperforated hymen, transverse vaginal septum, and vaginal agenesis are other anatomical disorders of female reproductive system that can lead to amenorrhea.[28]
Genetics
The major genes in amenorrhea
| Groups | Gene | Other name(s) | OMIM number | Chromosome | Function | Other related disorders |
|---|---|---|---|---|---|---|
| Kallmann syndrome
and Isolated hypogonadotropic hypogonadism[29] |
KAL1 | KAL1, anosmin-1 | 308700 | Xp22.3 |
|
|
| FGFR1 | KAL2 | 136350 | 8q12 |
|
| |
| PROKR2 | KAL3 | 607123 | 20p13 |
|
||
| PROK2 | KAL4 | 607002 | 3p21.1 | |||
| CHD7 | KAL5 | 608892 | 8q12.1 |
|
| |
| FGF8 | KAL6 | 600483 | 10q24 |
|
| |
| GPR54 | KISS1R | 604161 | 19p13.3 |
|
- | |
| KISS1 | KISS1, kisspeptin1 | 603286 | 1q32 |
|
- | |
| HS6ST1 | - | 604846 | 2q21 |
|
- | |
| TAC3 | NKB | 162330 | 12q13–q21 |
|
||
| TACR3 | NK3R | 152332 | 4q25 | |||
| GnRH1 | - | 152760 | 8p21–8p11.2 |
|
| |
| GnRHR | - | 138850 | 4q21.2 |
|
||
| NELF | - | 608137 | 9q34.3 | - | ||
| EBF2 | - | 609934 | 8p21.2 |
|
- | |
| HPG axis development | DAX1 | NR0B | 300473 | Xp21.2 |
|
|
| SF-1 | NR5A1 | 184757 | 9q33.3 |
|
||
| HESX-1 | RPX | 601802 | 3p14.3 |
|
| |
| LHX3 | LIM3 | 600577 | 9q34.3 |
|
| |
| PROP-1 | - | 601538 | 5q35.3 |
|
| |
| Obesity related
hypogonadotropic hypogonadism |
LEP | OB | 164160 | 7q32.1 |
|
|
| LEPR | OBR | 601007 | 1p31.3 | |||
| PC1 | NEC1 | 162150 | 5q15 |
|
| |
|
Abbreviations (alphabetic): | ||||||
Kisspeptin system (KISS1R and KISS1)
- The GPR54 gene, also called KISS1R, with Online Mendelian Inheritance in Man (OMIM) number of 604161 is on chromosome 19p13.3. The KISS1 gene, also known as kisspeptin1, with OMIM number of 603286 is on chromosome 1q32.
- Kisspeptin and related G-protein coupled receptor (KISS1R or GPR54) have key roles in the regulation of GnRH secretion. The GnRH secretion has to be pulsatile to stimulate gonadotropins. Kisspeptins are encoded by KISS1 gene, neuropeptides secreted from hypothalamus nuclei. It has been observed that patients with idiopathic hypogonadotropic hypogonadism have KISS1 receptor (GPR54) inactivating gene mutations.[30][31]
- By the time of onset of puberty, the KISS1 genes become activated through neuroanatomical and functional changes from environmental triggers, critical for brain sexual maturation and hypothalamic–pituitary–gonadal axis (HPG axis) activation with pulsatile GnRH.[32]
- Along HPG axis neurons, gamma-aminobutyric acid is inhibitory and glutamate is excitatory neurotransmitters. In related KNDy neurons in arcuate nucleus, the materials secreted are included kisspeptin, neurokinin B, and dynorphin A. Before puberty begins, inhibitory dynorphin A is the dominant element; decreased by stimulatory effect of neurokinin B, when puberty started. Conclusively, kisspeptin and GnRH/LH are increased.[33]
Kallmann syndrome 1 (KAL1)
- The KAL1 gene, also called anosmin-1, with OMIM number of 308700 is on chromosome Xp22.3, and encodes an extracellular matrix glycoprotein.
- Anosmin-1 is expressed at five weeks of gestation in forebrain near olfactory bulbs and stimulate the afferent fibers projections around it.[34]
- X-linked Kallmann syndrome is directly associated with KAL1 deletion. It is assumed to result in an absence of olfactory fibers along with disrupted migration of GnRH neurons, that are supposed to from migrated olfactory placode.[35]
- Male patient with KAL1 mutation would have central hypogonadism and anosmia/hyposmia. Additionally, the more diseases are assumed to relate with KAL1 gene, such as midline facial defects (cleft lip and/or cleft palate), short metacarpals, renal agenesis, sensorineural hearing loss, bimanual synkinesis, oculomotor abnormalities, and cerebellar ataxia.[36]
Fibroblast growth factor receptor 1 and fibroblast growth factor 8 (FGFR1 and FGF8)
- The FGFR1 gene, also called KAL2, with OMIM number of 136350 is on chromosome 8q12, encode a receptor tyrosine kinase protein. The FGF8 gene, also called KAL6, is on chromosome 10q24.
- FGFR1 pathway is assumed to be the main role in embryogenesis, homeostasis, and wound healing. FGF8 plays a critical role in the primary generation of neural tissue.[37]
- On the other hand, interaction between FGFR1, FGF8, and heparan sulfate helps the olfactory bulb to become differentiated and developed and also facilitates GnRH neurons in migration and function.[38]
- Dominant deletion mutation of FGFR1 gene is associated with a 30% decrease in hypothalamic GnRH neurons.[39] Other defects related to FGFR1 includes cleft palate or lip, dental agenesis and bimanual synkinesis.[36]
Heparan sulfate 6-O-sulphotransferase 1 (HS6ST1)
- The HS6ST1 gene with OMIM number of 604846 is on chromosome 2q21 has been found to be mutated in hypogonadism.[40]
- The modifications of heparan sulfate polysaccharides in extracellular matrix have some role in FGFR-FGF and also anosmin1-cell membrane interactions.[41][42]
- This gene may be mutated in both Kallmann syndrome and idiopathic hypogonadism resulting in various courses, disruption pf frequency of GnRH secretion, and/or GnRH deficiencies.[40]
Prokineticin 2 and prokineticin 2 receptor (PROK2 and PROKR2)
- The PROK2 and PROKR2 genes, also called KAL4 and KAL3, with OMIM numbers of 607002 and 607123 are on chromosomes 3p21.1 and 20p13, respectively. They are believed to be a cause of Kallmann syndrome.
- PROKR2 is a G protein coupled receptor (GPCR), has a major role in olfactory bulb development; and its mutation may lead to severe gonadal atrophy.[43]
- In prokineticin system, there are two receptors (PROKR1 and PROKR2) and two ligands (PROK1 and PROK2). PROK1 and its receptor (PROKR1) have some role in gastrointestinal system motility. PROK2 and PROKR2 are parts of neuroendocrine system, located in arcuate nucleus, olfactory tract, and suprachiasmatic nucleus.[44]
- The mutated versions of PROK2 and PROKR2 could lead to decrease GnRH production and hypogonadism. Other disorders caused by PROK2 and PROKR2 mutations include fibrous dysplasia, sleep disorder, severe obesity, synkinesis, and epilepsy.[45]
Tachykinin 3 and tachykinin 3 receptor (TAC3 and TACR3)
- The TAC3 and TACR3 genes, also called neurokinin B (NKB) and neurokinin 3 receptor (NK3R), with OMIM numbers of 162330 and 152332, are on chromosomes 12q13–q21 and 4q25, respectively.[46]
- It is postulated that the normal function of TAC3/TACR3 system is necessary for an intact HPG axis and its development during puberty. On the other hand, TAC3/TACR3 system disturbance may cause micropenis and cryptorchidism in males, showing the major role of TAC3/TACR3 in fetal gonadotropins secretion.[47]
- TACR3 encoded protein (NK3R) is GPCR, initially produced in central nervous system. The major mechanism, through which the mutated gene may lead to neuroendocrine disturbance and delayed puberty, is not yet discovered completely.[48]
- TAC3 encoded protein (NKB) is produced in arcuate nucleus of hypothalamus and play an important role in GnRH secretion. Kisspeptin is also produced and secreted in arcuate nucleus, whereas, both of them are inhibited by estrogen. It may be considered that kisspeptin and NKB have identical roles in diverting negative feedback from sex hormones to GnRH. Their mutation has been shown to be related with hypogonadism.
Gonadotropin releasing hormone and its receptor (GnRH1 and GnRHR)
- The GnRH1 and GnRHR genes with OMIM numbers of 152760 and 138850 are on chromosomes 8p21–8p11.2 and 4q21.2, respectively.[49]
- In HPG axis, GnRH is one of the most effective elements; therefore, a defect could directly influence the axis and slow down the progress.
- The GnRHR gene is also responsible for gonadal normal functions and its mutation could lead to hypogonadism and delayed puberty. The mutation in GnRHR has also been associated with conditions such as atrophic gonads along with low LH/FSH and sex hormones, sexual puberty disturbance, inability to conceive, and failure to impact from exogenous GnRH.[50]
- GnRH1 and GnRHR genes have variable expression and cause a spectrum of symptoms, from fertile eunuch syndrome and partial idiopathic hypogonadotropic hypogonadism to complete GnRH resistance (i.e., characterized by cryptorchidism), microphallus, very low LH/FSH, and delayed puberty.[51]
- The other disorders that have been found to be related to GnRH mutation are tooth maturation and biomineralization.[52]
Chromodomain helicase DNA-binding protein 7 (CHD7)
- The CHD7 gene, also called as KAL5, with OMIM number of 608892 is on chromosome 8q12.1.
- CHD7 gene mutation results in autosomal dominant CHARGE syndrome, which is a combination of hypogonadism and Kallmann syndrome, and includes:[53]
- Coloboma
- Heart anomalies
- Choanal Atresia
- Retardation
- Genital anomalies
- Ear anomalies
- Screening for CHD7 gene mutation may be done in patients with hypogonadism or Kallmann syndrome with specific features such as semicircular canal hypoplasia or aplasia, dysmorphic ears, and deafness.
Nasal embryonic LH-releasing hormone factor (NELF)
- The NELF gene with OMIM number of 608137 is on chromosome 9q34.3; present mostly in nervous tissues specifically during fetal development and may be found in olfactory bulb and pituitary LH releasing cells.
- The most common function is in olfactory axons and GnRH neurons, before and during neuron migration in the developmental process.[54]
Early B-cell factor 2 (EBF2)
- The EBF2 gene with OMIM number of 609934 is on chromosome 8p21.2; mostly expressed in mice osteoblasts and osteoclast cells.[55]
- EBF2 gene plays an effective role in HPG axis. Mutation in EBF2 gene can result in disruption of HPG axis, leading to secondary hypogonadism.[56]
DSS-AHC on the X-chromosome 1 (DAX1)
- The DAX1 gene, also called nuclear receptor 0B (NR0B), with OMIM number of 300473 is on chromosome Xp21.2, and expressed in all members of HPG axis (hypothalamus, pituitary, and gonads).[57]
- During the spermatogenesis and steroidogenesis, both sertoli and leydig cells have increased expression of DAX1 gene. It is assumed that during puberty, the peak expression of DAX1 is observed.[58]
- Other disease that can be caused by DAX1 mutation is congenital adrenal cortex hypoplasia.[59]
Steroidogenic factor 1 (SF1)
- The SF1 gene, also called nuclear receptor 5A1 (NR5A1), with OMIM number of 184757 is on chromosome 9q33.3, has some role in reproduction, steroidogenesis, and sexual differentiation.
- It is mainly expressed in sertoli and leydig cells, and plays an important role in steroidogenesis and spermatogenesis. The SF1 is believed to have an increase in expression from childhood until adolescence, and is dominantly expressed by leydig cells in puberty.[58]
- Other diseases that may be caused by SF1 mutation include male pseudohermaphroditism, Denys-Drash syndrome, and also hypospadias.[60]
Homeobox gene 1 (HESX1)
- The HESX1 gene, also called Rathke pouch homeobox (RPX), with OMIM number of 601802 is on chromosome 3p14.3, initially expressed during embryogenesis and help the formation of Rathke pouch and anterior pituitary.[61]
- HESX1 gene has an important role in pituitary development and midfacial differentiation. Mutation may lead to pituitary hypoplasia and decreased level of all anterior pituitary hormones.[62]
- Other disorders resulting from HESX1 mutation include septo optic dysplasia, reduced prosencephalon, anophthalmia, microphthalmia, defective olfactory development, Rathke pouch bifurcations, and abnormalities in the corpus callosum, hippocampus, and septum pellucidum.[61]
LIM homeobox gene 3 (LHX3)
- The LHX3 gene, also called LIM3, with OMIM number of 600577 is on chromosome 9q34.3, mainly expressed in developing anterior pituitary gland.[63]
- LHX3 gene function is important in development of pituitary gland and pituitary hormones secretion. Mutation in the LHX3 gene may result in combined pituitary hormone deficiency (CPHD).[64]
- LHX3 gene mutation may also result in neonatal hypoglycemia, short neck with limited rotation, mild sensorineural hearing loss, skin laxity, and skeletal abnormalities.[63]
PROP paired-like homeobox 1 (PROP1)
- The PROP1 gene with OMIM number of 601538 is on chromosome 5q35.3, with a role in developing anterior pituitary gland and associated cells such as gonadotrophs, thyrotrophs, somatotrophs, and lactotrophs.[65]
- Mutated PROP1 gene can lead to deficiency of LH, FSH, GH, TSH, and prolactin. Decreased level of LH and FSH may also delay or inhibit the onset of puberty.[66]
- Pituitary hormones have a vital role in regulating other endocrine organs via TRH, ACTH, FSH or LH and a mutation in PROP1 gene can lead to thyroid dysfunctions, growth retardation, and libido/lactation problems.
Leptin and leptin receptor (LEP and LEPR)
- The LEP and LEPR genes, also called OB and OBR, with OMIM numbers of 164160 and 601007 are on chromosomes 7q32.1 and 1p31.3, respectively; with a major role in modulation of body weight.
- These genes are believed to carry the message of onset of puberty. Recent studies have shown that recombinant leptin injection in female mice may result in puberty and cure their maturation (secondary sexual characteristics) problems.[67]
- It has been observed that leptin levels increase by 50% just before the onset of puberty and during puberty.[68]
- Mutation in LEP and LEPR genes may result in dysfunctional hematopoiesis, angiogenesis, wound healing, and the immune or inflammatory response.
Proprotein convertase 1 (PC1)
- The PC1 gene, also known as neuroendocrine convertase 1 (NEC1), with OMIM number of 162150 is on chromosome 5q15, and regulates neuroendocrine pathway.
- PC1 gene has a dominant role in proopiomelanocortin (POMC) cleavage. PC1 gene also has a role in processing proinsulin and proglucagon in pancreas.[69]
- Recent studies have shown that PC1 gene mutation and hypogonadotropic hypogonadism may result in extreme childhood obesity, abnormal glucose homeostasis, hypocortisolism, elevated plasma proinsulin, and POMC concentrations.[70]
Makorin RING-finger protein 3 (MKRN3)
- Newly discovered MKRN3 gene has a role in ubiquitination and cell signaling. The gene family proteins are majorly expressed in fetal brain during development, especially in arcuate nucleus.
- The process of gene amplification is on its peak after birth, which gradually declines with time, and finally rises again with onset of puberty. Thus MKRN3 gene is believed to be one of the factors in onset of puberty, along with kisspeptins and neurokinin B.[71]
Estrogen receptor α (ESR1)
- Estrogen receptor mutations are very rare, and were reported in a case report of delayed puberty.[72]
- Estradiol promotes breast maturation and provides negative feedback to hypothalamus and pituitary, by means of estrogen receptor α (encoded by ESR1 gene).[73]
- Female mice with mutated ESR1 gene have hypoplastic uterus with hemorrhage, multicystic ovary without corpus luteum; which is make them infertile.[74]
Associated Conditions
The associated conditions that are related to amenorrhea, are as following:[75]
Gross Pathology

- On gross pathology, normal endometrium in proliferative or luteal phases are characteristic findings of amenorrhea.
- In cases of amenorrhea which are secondary to other causes, the related gross pathology would be seen.
- Craniopharyngioma on gross pathology presents as cystic mass filled with motor oil-like fluid.[76]
Microscopic Pathology
- On microscopic histopathological analysis, craniopharyngioma as a cause of amenorrhea will have the following features:
- Trabecular squamous epithelium surrounded by palisaded columnar epithelium
- Small-to-medium sized cells with moderate amount of basophilic cytoplasm
- Bland nuclei
- Calcifications
- On microscopic histopathological analysis, pituitary adenoma as a cause of amenorrhea will have the following features:
- Loss of fibrous stroma
- Nested cells of normal anterior pituitary (based on the type of adenoma)
-
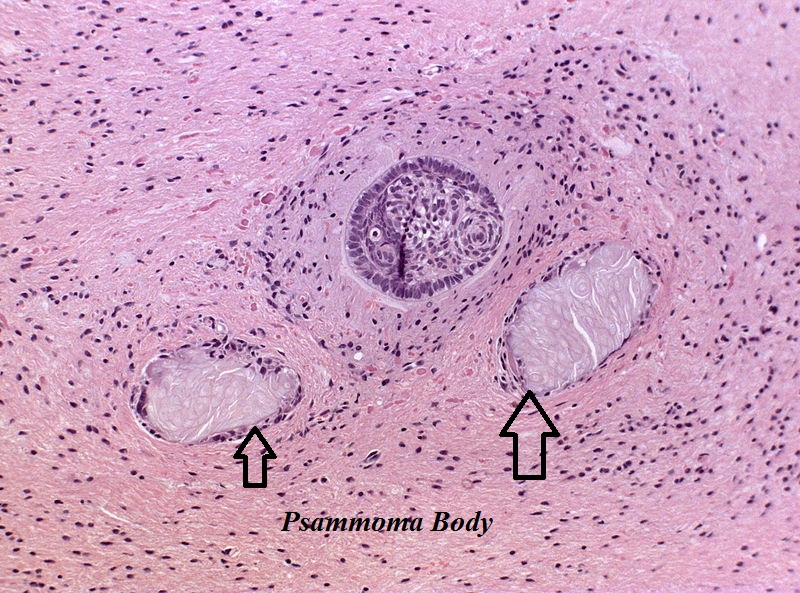
Craniopharyngioma; with psammoma bodies - by Jensflorian source: Librepathology
-
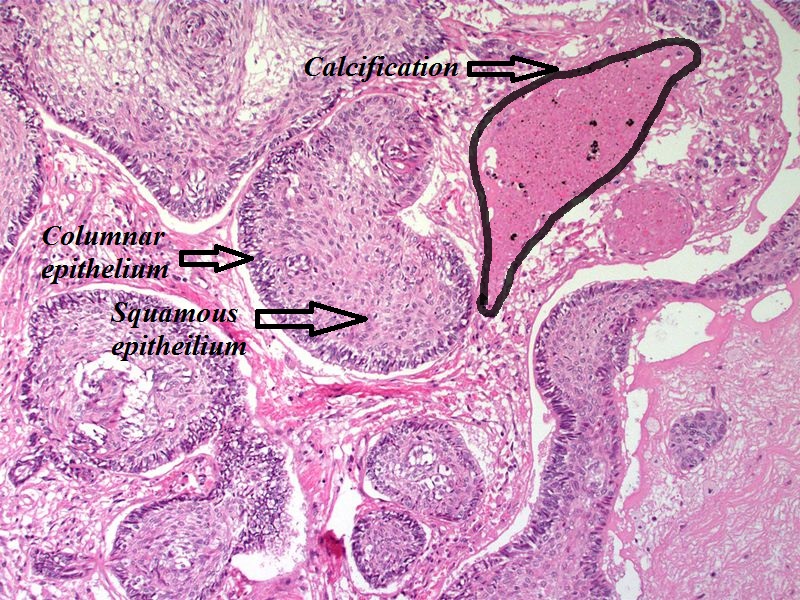
Craniopharyngioma; with calcification and two types of epithelium - by Sarahkayb source: Librepathology
-
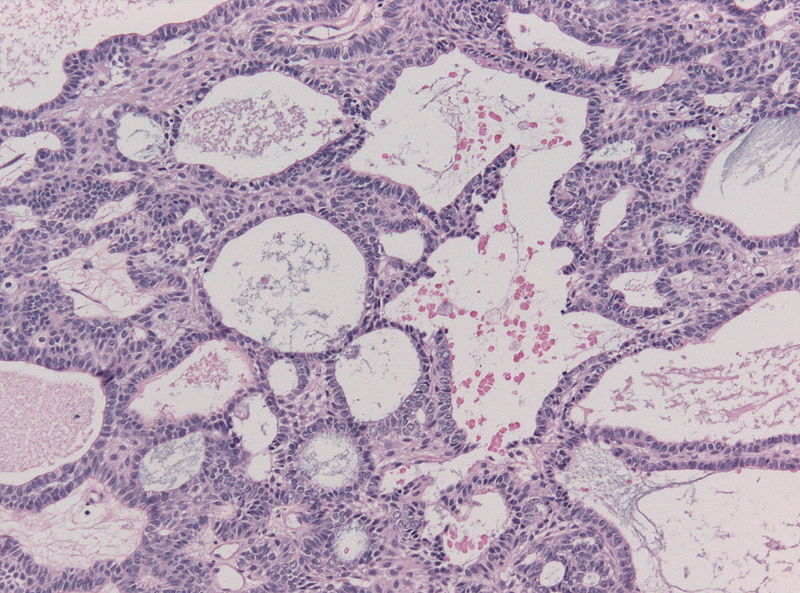
Craniopharyngioma; multicystic texture - by Jensflorian source: Librepathology
-
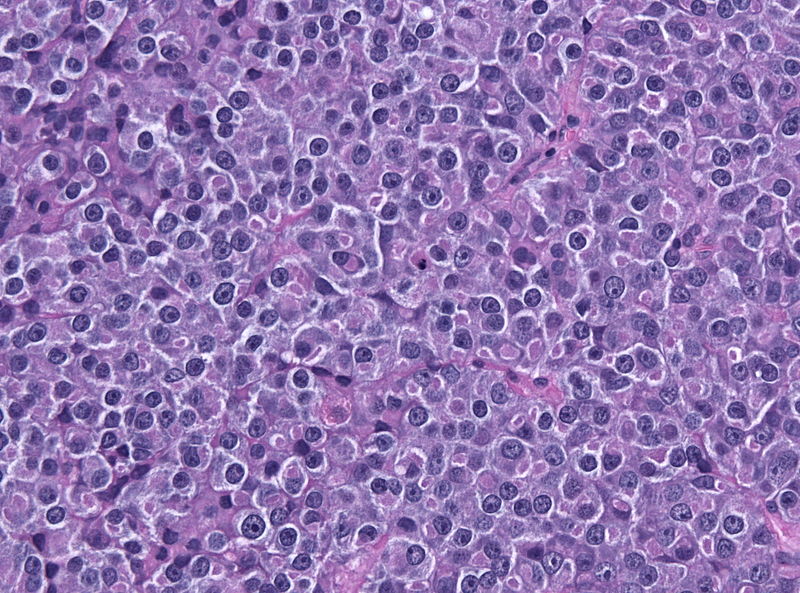
Pituitary adenoma of lactotroph cells (prolactin producing) - by Jensflorian source: Librepathology
-
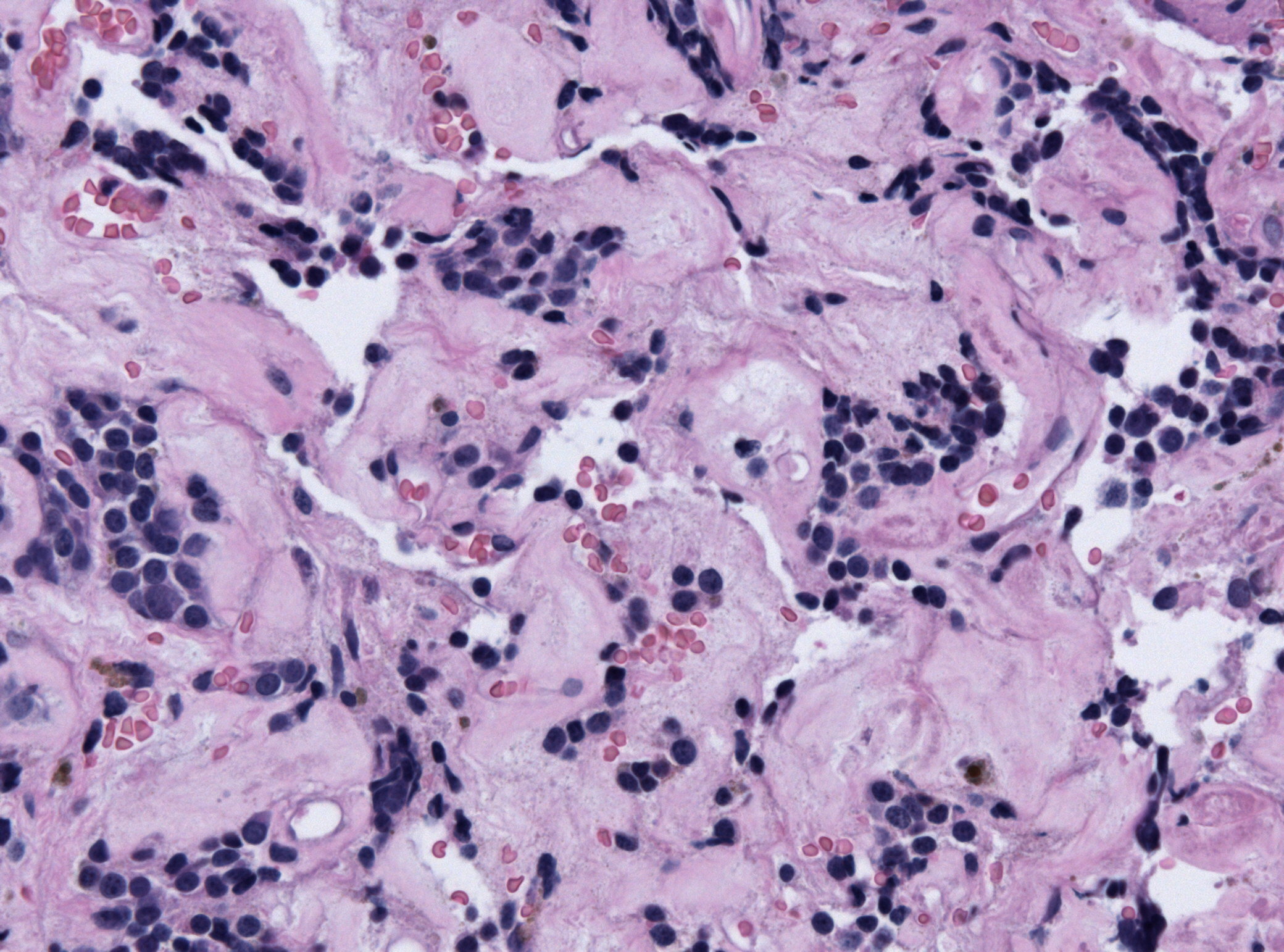
Pituitary adenoma of lactotroph cells (prolactin producing) regression after treatment - by Jensflorian source: Librepathology
-
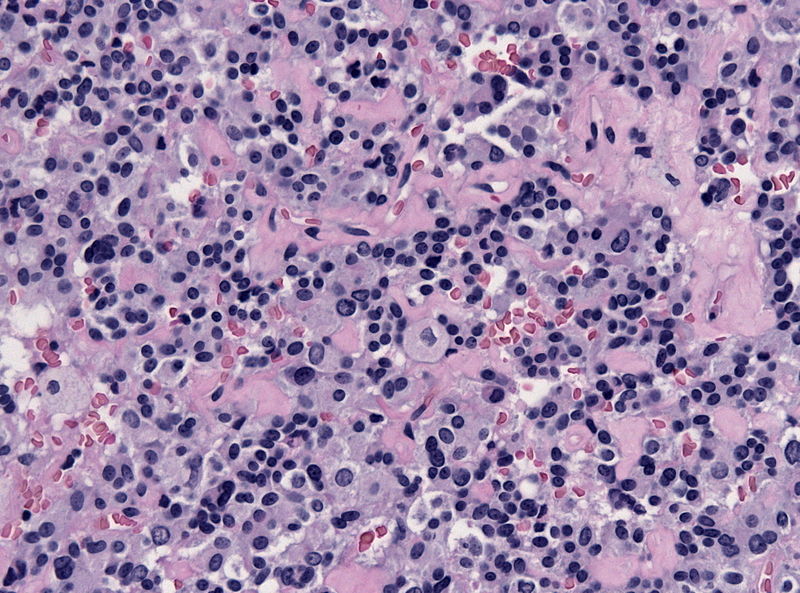
Pituitary adenoma of thyrotroph cells (TSH producing) - by Jensflorian source: Librepathology






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- ↑ Chumlea WC, Schubert CM, Roche AF, Kulin HE, Lee PA, Himes JH, Sun SS (2003). "Age at menarche and racial comparisons in US girls". Pediatrics. 111 (1): 110–3. PMID 12509562.
- ↑ "Menstruation in Girls and Adolescents: Using the Menstrual Cycle as a Vital Sign - ACOG".
- ↑ 3.0 3.1 Golden NH, Carlson JL (2008). "The pathophysiology of amenorrhea in the adolescent". Ann. N. Y. Acad. Sci. 1135: 163–78. doi:10.1196/annals.1429.014. PMID 18574222.
- ↑ Apter D (1997). "Development of the hypothalamic-pituitary-ovarian axis". Ann. N. Y. Acad. Sci. 816: 9–21. PMID 9238251.
- ↑ Boyar RM, Rosenfeld RS, Kapen S, Finkelstein JW, Roffwarg HP, Weitzman ED, Hellman L (1974). "Human puberty. Simultaneous augmented secretion of luteinizing hormone and testosterone during sleep". J. Clin. Invest. 54 (3): 609–18. doi:10.1172/JCI107798. PMC 301594. PMID 4852310.
- ↑ Wiksten-Almströmer M, Hirschberg AL, Hagenfeldt K (2007). "Menstrual disorders and associated factors among adolescent girls visiting a youth clinic". Acta Obstet Gynecol Scand. 86 (1): 65–72. doi:10.1080/00016340601034970. PMID 17230292.
- ↑ Perkins RB, Hall JE, Martin KA (2001). "Aetiology, previous menstrual function and patterns of neuro-endocrine disturbance as prognostic indicators in hypothalamic amenorrhoea". Hum. Reprod. 16 (10): 2198–205. PMID 11574516.
- ↑ Hebebrand J, Muller TD, Holtkamp K, Herpertz-Dahlmann B (2007). "The role of leptin in anorexia nervosa: clinical implications". Mol. Psychiatry. 12 (1): 23–35. doi:10.1038/sj.mp.4001909. PMID 17060920.
- ↑ Grinspoon S, Gulick T, Askari H, Landt M, Lee K, Anderson E, Ma Z, Vignati L, Bowsher R, Herzog D, Klibanski A (1996). "Serum leptin levels in women with anorexia nervosa". J. Clin. Endocrinol. Metab. 81 (11): 3861–3. doi:10.1210/jcem.81.11.8923829. PMID 8923829.
- ↑ Haas V, Onur S, Paul T, Nutzinger DO, Bosy-Westphal A, Hauer M, Brabant G, Klein H, Müller MJ (2005). "Leptin and body weight regulation in patients with anorexia nervosa before and during weight recovery". Am. J. Clin. Nutr. 81 (4): 889–96. PMID 15817868.
- ↑ Misra M, Miller KK, Almazan C, Ramaswamy K, Aggarwal A, Herzog DB, Neubauer G, Breu J, Klibanski A (2004). "Hormonal and body composition predictors of soluble leptin receptor, leptin, and free leptin index in adolescent girls with anorexia nervosa and controls and relation to insulin sensitivity". J. Clin. Endocrinol. Metab. 89 (7): 3486–95. doi:10.1210/jc.2003-032251. PMID 15240636.
- ↑ Katzman DK, Golden NH, Neumark-Sztainer D, Yager J, Strober M (2000). "From prevention to prognosis: clinical research update on adolescent eating disorders". Pediatr. Res. 47 (6): 709–12. PMID 10832726.
- ↑ Thong FS, McLean C, Graham TE (2000). "Plasma leptin in female athletes: relationship with body fat, reproductive, nutritional, and endocrine factors". J. Appl. Physiol. 88 (6): 2037–44. PMID 10846016.
- ↑ Weimann E, Blum WF, Witzel C, Schwidergall S, Böhles HJ (1999). "Hypoleptinemia in female and male elite gymnasts". Eur. J. Clin. Invest. 29 (10): 853–60. PMID 10583427.
- ↑ Welt, Corrine K.; Chan, Jean L.; Bullen, John; Murphy, Robyn; Smith, Patricia; DePaoli, Alex M.; Karalis, Aspasia; Mantzoros, Christos S. (2004). "Recombinant Human Leptin in Women with Hypothalamic Amenorrhea". New England Journal of Medicine. 351 (10): 987–997. doi:10.1056/NEJMoa040388. ISSN 0028-4793.
- ↑ Wieck A, Haddad PM (2003). "Antipsychotic-induced hyperprolactinaemia in women: pathophysiology, severity and consequences. Selective literature review". Br J Psychiatry. 182: 199–204. PMID 12611781.
- ↑ Magiakou MA, Mastorakos G, Webster E, Chrousos GP (1997). "The hypothalamic-pituitary-adrenal axis and the female reproductive system". Ann. N. Y. Acad. Sci. 816: 42–56. PMID 9238254.
- ↑ Seminara SB, Hayes FJ, Crowley WF (1998). "Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations". Endocr. Rev. 19 (5): 521–39. doi:10.1210/edrv.19.5.0344. PMID 9793755.
- ↑ Karavitaki N, Cudlip S, Adams CB, Wass JA (2006). "Craniopharyngiomas". Endocr. Rev. 27 (4): 371–97. doi:10.1210/er.2006-0002. PMID 16543382.
- ↑ Koutras DA (1997). "Disturbances of menstruation in thyroid disease". Ann. N. Y. Acad. Sci. 816: 280–4. PMID 9238278.
- ↑ Poppe K, Velkeniers B, Glinoer D (2007). "Thyroid disease and female reproduction". Clin. Endocrinol. (Oxf). 66 (3): 309–21. doi:10.1111/j.1365-2265.2007.02752.x. PMID 17302862.
- ↑ Speiser, Phyllis W.; White, Perrin C. (2003). "Congenital Adrenal Hyperplasia". New England Journal of Medicine. 349 (8): 776–788. doi:10.1056/NEJMra021561. ISSN 0028-4793.
- ↑ Gilling-Smith C, Story H, Rogers V, Franks S (1997). "Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome". Clin. Endocrinol. (Oxf). 47 (1): 93–9. PMID 9302378.
- ↑ Ehrmann DA, Barnes RB, Rosenfield RL (1995). "Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion". Endocr. Rev. 16 (3): 322–53. doi:10.1210/edrv-16-3-322. PMID 7671850.
- ↑ Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC (1998). "Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone". J. Clin. Endocrinol. Metab. 83 (2): 582–90. doi:10.1210/jcem.83.2.4604. PMID 9467578.
- ↑ Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R (1981). "Hypergonadotropic hypogonadism in female patients with galactosemia". N. Engl. J. Med. 304 (17): 994–8. doi:10.1056/NEJM198104233041702. PMID 6782485.
- ↑ Varner RE, Younger JB, Blackwell RE (1985). "Müllerian dysgenesis". J Reprod Med. 30 (6): 443–50. PMID 4020785.
- ↑ Edmonds DK (2003). "Congenital malformations of the genital tract and their management". Best Pract Res Clin Obstet Gynaecol. 17 (1): 19–40. PMID 12758224.
- ↑ Bonomi, Marco; Libri, Domenico Vladimiro; Guizzardi, Fabiana; Guarducci, Elena; Maiolo, Elisabetta; Pignatti, Elisa; Asci, Roberta; Persani, Luca (2011). "New understandings of the genetic basis of isolated idiopathic central hypogonadism". Asian Journal of Andrology. 14 (1): 49–56. doi:10.1038/aja.2011.68. ISSN 1008-682X.
- ↑ de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E (2003). "Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54". Proc. Natl. Acad. Sci. U.S.A. 100 (19): 10972–6. doi:10.1073/pnas.1834399100. PMC 196911. PMID 12944565.
- ↑ Seminara, Stephanie B.; Messager, Sophie; Chatzidaki, Emmanouella E.; Thresher, Rosemary R.; Acierno, James S.; Shagoury, Jenna K.; Bo-Abbas, Yousef; Kuohung, Wendy; Schwinof, Kristine M.; Hendrick, Alan G.; Zahn, Dirk; Dixon, John; Kaiser, Ursula B.; Slaugenhaupt, Susan A.; Gusella, James F.; O'Rahilly, Stephen; Carlton, Mark B.L.; Crowley, William F.; Aparicio, Samuel A.J.R.; Colledge, William H. (2003). "TheGPR54Gene as a Regulator of Puberty". New England Journal of Medicine. 349 (17): 1614–1627. doi:10.1056/NEJMoa035322. ISSN 0028-4793.
- ↑ Kaur KK, Allahbadia G, Singh M (2012). "Kisspeptins in human reproduction-future therapeutic potential". J Assist Reprod Genet. 29 (10): 999–1011. doi:10.1007/s10815-012-9856-1. PMC 3492584. PMID 23015158.
- ↑ Uenoyama, Yoshihisa; Tsukamura, Hiroko; Maeda, Kei-ichiro (2014). "KNDy neuron as a gatekeeper of puberty onset". Journal of Obstetrics and Gynaecology Research. 40 (6): 1518–1526. doi:10.1111/jog.12398. ISSN 1341-8076.
- ↑ Hardelin JP, Julliard AK, Moniot B, Soussi-Yanicostas N, Verney C, Schwanzel-Fukuda M, Ayer-Le Lievre C, Petit C (1999). "Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome". Dev. Dyn. 215 (1): 26–44. doi:10.1002/(SICI)1097-0177(199905)215:1<26::AID-DVDY4>3.0.CO;2-D. PMID 10340754.
- ↑ Schwanzel-Fukuda M, Bick D, Pfaff DW (1989). "Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome". Brain Res. Mol. Brain Res. 6 (4): 311–26. PMID 2687610.
- ↑ 36.0 36.1 Trarbach EB, Silveira LG, Latronico AC (2007). "Genetic insights into human isolated gonadotropin deficiency". Pituitary. 10 (4): 381–91. doi:10.1007/s11102-007-0061-7. PMID 17624596.
- ↑ González-Martínez D, Kim SH, Hu Y, Guimond S, Schofield J, Winyard P, Vannelli GB, Turnbull J, Bouloux PM (2004). "Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism". J. Neurosci. 24 (46): 10384–92. doi:10.1523/JNEUROSCI.3400-04.2004. PMID 15548653.
- ↑ Hébert JM, Lin M, Partanen J, Rossant J, McConnell SK (2003). "FGF signaling through FGFR1 is required for olfactory bulb morphogenesis". Development. 130 (6): 1101–11. PMID 12571102.
- ↑ Tsai PS, Moenter SM, Postigo HR, El Majdoubi M, Pak TR, Gill JC, Paruthiyil S, Werner S, Weiner RI (2005). "Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population". Mol. Endocrinol. 19 (1): 225–36. doi:10.1210/me.2004-0330. PMID 15459253.
- ↑ 40.0 40.1 Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Seminara SB, Hughes V, Van Vliet G, Van Uum S, Crowley WF, Habuchi H, Kimata K, Pitteloud N, Bülow HE (2011). "Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism". Proc. Natl. Acad. Sci. U.S.A. 108 (28): 11524–9. doi:10.1073/pnas.1102284108. PMC 3136273. PMID 21700882.
- ↑ Ibrahimi OA, Zhang F, Hrstka SC, Mohammadi M, Linhardt RJ (2004). "Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly". Biochemistry. 43 (16): 4724–30. doi:10.1021/bi0352320. PMID 15096041.
- ↑ Hudson ML, Kinnunen T, Cinar HN, Chisholm AD (2006). "C. elegans Kallmann syndrome protein KAL-1 interacts with syndecan and glypican to regulate neuronal cell migrations". Dev. Biol. 294 (2): 352–65. doi:10.1016/j.ydbio.2006.02.036. PMID 16677626.
- ↑ Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y (2006). "Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2". Proc. Natl. Acad. Sci. U.S.A. 103 (11): 4140–5. doi:10.1073/pnas.0508881103. PMC 1449660. PMID 16537498.
- ↑ Li M, Bullock CM, Knauer DJ, Ehlert FJ, Zhou QY (2001). "Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle". Mol. Pharmacol. 59 (4): 692–8. PMID 11259612.
- ↑ Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Raivio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley WF, Zhou QY, Pitteloud N (2008). "Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum". J. Clin. Endocrinol. Metab. 93 (9): 3551–9. doi:10.1210/jc.2007-2654. PMC 2567850. PMID 18559922.
- ↑ Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK (2009). "TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction". Nat. Genet. 41 (3): 354–358. doi:10.1038/ng.306. PMC 4312696. PMID 19079066.
- ↑ Pinto FM, Almeida TA, Hernandez M, Devillier P, Advenier C, Candenas ML (2004). "mRNA expression of tachykinins and tachykinin receptors in different human tissues". Eur. J. Pharmacol. 494 (2–3): 233–9. doi:10.1016/j.ejphar.2004.05.016. PMID 15212980.
- ↑ Semple RK, Topaloglu AK (2010). "The recent genetics of hypogonadotrophic hypogonadism - novel insights and new questions". Clin. Endocrinol. (Oxf). 72 (4): 427–35. doi:10.1111/j.1365-2265.2009.03687.x. PMID 19719764.
- ↑ Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J (2009). "Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation". N. Engl. J. Med. 360 (26): 2742–8. doi:10.1056/NEJMoa0900136. PMID 19535795.
- ↑ Wu S, Wilson MD, Busby ER, Isaac ER, Sherwood NM (2010). "Disruption of the single copy gonadotropin-releasing hormone receptor in mice by gene trap: severe reduction of reproductive organs and functions in developing and adult mice". Endocrinology. 151 (3): 1142–52. doi:10.1210/en.2009-0598. PMID 20068010.
- ↑ Silveira LF, MacColl GS, Bouloux PM (2002). "Hypogonadotropic hypogonadism". Semin. Reprod. Med. 20 (4): 327–38. doi:10.1055/s-2002-36707. PMID 12536356.
- ↑ Tiong J, Locastro T, Wray S (2007). "Gonadotropin-releasing hormone-1 (GnRH-1) is involved in tooth maturation and biomineralization". Dev. Dyn. 236 (11): 2980–92. doi:10.1002/dvdy.21332. PMID 17948256.
- ↑ Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC (2008). "Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome". Am. J. Hum. Genet. 83 (4): 511–9. doi:10.1016/j.ajhg.2008.09.005. PMC 2561938. PMID 18834967.
- ↑ Kramer PR, Wray S (2000). "Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons". Genes Dev. 14 (14): 1824–34. PMC 316793. PMID 10898796.
- ↑ Corradi A, Croci L, Broccoli V, Zecchini S, Previtali S, Wurst W, Amadio S, Maggi R, Quattrini A, Consalez GG (2003). "Hypogonadotropic hypogonadism and peripheral neuropathy in Ebf2-null mice". Development. 130 (2): 401–10. PMID 12466206.
- ↑ Trarbach EB, Baptista MT, Garmes HM, Hackel C (2005). "Molecular analysis of KAL-1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients". J. Endocrinol. 187 (3): 361–8. doi:10.1677/joe.1.06103. PMID 16423815.
- ↑ Guo W, Burris TP, McCabe ER (1995). "Expression of DAX-1, the gene responsible for X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism, in the hypothalamic-pituitary-adrenal/gonadal axis". Biochem. Mol. Med. 56 (1): 8–13. PMID 8593542.
- ↑ 58.0 58.1 Kojima Y, Sasaki S, Hayashi Y, Umemoto Y, Morohashi K, Kohri K (2006). "Role of transcription factors Ad4bp/SF-1 and DAX-1 in steroidogenesis and spermatogenesis in human testicular development and idiopathic azoospermia". Int. J. Urol. 13 (6): 785–93. doi:10.1111/j.1442-2042.2006.01403.x. PMID 16834661.
- ↑ Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER (1994). "An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita". Nature. 372 (6507): 635–41. doi:10.1038/372635a0. PMID 7990953.
- ↑ Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan JN, Hammer GD, Ingraham HA (1998). "Wilms' tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression". Cell. 93 (3): 445–54. PMID 9590178.
- ↑ 61.0 61.1 Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Mårtensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC (1998). "Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse". Nat. Genet. 19 (2): 125–33. doi:10.1038/477. PMID 9620767.
- ↑ Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS (2001). "Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia". Hum. Mol. Genet. 10 (1): 39–45. PMID 11136712.
- ↑ 63.0 63.1 Rajab A, Kelberman D, de Castro SC, Biebermann H, Shaikh H, Pearce K, Hall CM, Shaikh G, Gerrelli D, Grueters A, Krude H, Dattani MT (2008). "Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss". Hum. Mol. Genet. 17 (14): 2150–9. doi:10.1093/hmg/ddn114. PMID 18407919.
- ↑ Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers A, Goossens M, Grüters A, Amselem S (2000). "Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency". Nat. Genet. 25 (2): 182–6. doi:10.1038/76041. PMID 10835633. Vancouver style error: initials (help)
- ↑ Duquesnoy P, Roy A, Dastot F, Ghali I, Teinturier C, Netchine I, Cacheux V, Hafez M, Salah N, Chaussain JL, Goossens M, Bougnères P, Amselem S (1998). "Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency". FEBS Lett. 437 (3): 216–20. PMID 9824293.
- ↑ Wu W, Cogan JD, Pfäffle RW, Dasen JS, Frisch H, O'Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA, Rosenfeld MG (1998). "Mutations in PROP1 cause familial combined pituitary hormone deficiency". Nat. Genet. 18 (2): 147–9. doi:10.1038/ng0298-147. PMID 9462743.
- ↑ Chehab FF, Lim ME, Lu R (1996). "Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin". Nat. Genet. 12 (3): 318–20. doi:10.1038/ng0396-318. PMID 8589726.
- ↑ Mantzoros CS, Flier JS, Rogol AD (1997). "A longitudinal assessment of hormonal and physical alterations during normal puberty in boys. V. Rising leptin levels may signal the onset of puberty". J. Clin. Endocrinol. Metab. 82 (4): 1066–70. doi:10.1210/jcem.82.4.3878. PMID 9100574.
- ↑ Jansen E, Ayoubi TA, Meulemans SM, Van de Ven WJ (1995). "Neuroendocrine-specific expression of the human prohormone convertase 1 gene. Hormonal regulation of transcription through distinct cAMP response elements". J. Biol. Chem. 270 (25): 15391–7. PMID 7797529.
- ↑ Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S (1997). "Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene". Nat. Genet. 16 (3): 303–6. doi:10.1038/ng0797-303. PMID 9207799.
- ↑ Hughes, Ieuan A. (2013). "Releasing the Brake on Puberty". New England Journal of Medicine. 368 (26): 2513–2515. doi:10.1056/NEJMe1306743. ISSN 0028-4793.
- ↑ Quaynor, Samuel D.; Stradtman, Earl W.; Kim, Hyung-Goo; Shen, Yiping; Chorich, Lynn P.; Schreihofer, Derek A.; Layman, Lawrence C. (2013). "Delayed Puberty and Estrogen Resistance in a Woman with Estrogen Receptor α Variant". New England Journal of Medicine. 369 (2): 164–171. doi:10.1056/NEJMoa1303611. ISSN 0028-4793.
- ↑ Christian CA, Glidewell-Kenney C, Jameson JL, Moenter SM (2008). "Classical estrogen receptor alpha signaling mediates negative and positive feedback on gonadotropin-releasing hormone neuron firing". Endocrinology. 149 (11): 5328–34. doi:10.1210/en.2008-0520. PMC 2584581. PMID 18635656.
- ↑ Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O (1993). "Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene". Proc. Natl. Acad. Sci. U.S.A. 90 (23): 11162–6. PMC 47942. PMID 8248223.
- ↑
- ↑ Fernandez-Miranda JC, Gardner PA, Snyderman CH, Devaney KO, Strojan P, Suárez C, Genden EM, Rinaldo A, Ferlito A (2012). "Craniopharyngioma: a pathologic, clinical, and surgical review". Head Neck. 34 (7): 1036–44. doi:10.1002/hed.21771. PMID 21584897.