Pheochromocytoma pathophysiology
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma pathophysiology |
|
Risk calculators and risk factors for Pheochromocytoma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
It is understood that pheochromocytoma is mediated by excessive secretion of catecholamines and subsequent stimulation of adrenergic receptors. It arises from the chromaffin cells of the adrenal medulla and sympathetic ganglia. The pathophysiology of pheochromocytoma does not depend on the histological subtype. Malignant and benign pheochromocytomas share the same biochemical and histological features. It may be sporadic or familial. All of these forms have genetic origin depending on a large number of genes, for example, VHL, SDH, NF1, RET genes. It is associated with conditions like MEN 2A syndrome, MEN 2B syndrome, VHL disease, and NF1.
Pathophysiology
Physiology
Pheochromocytoma is not associated with normal physiology.
Pathology
- It is understood that pheochromocytoma is the is mediated by excessive secretion of catecholamines and subsequent stimulation of adrenergic receptors.
- Commonly secreted catecholamines include norepinephrine (predominant) and epinephrine. Some tumors may also secrete dopamine. [1]
- Excessive secretion of catecholamines may be either continuous or intermittent.
- Pheochromocytoma is a tumor which arises from the chromaffin cells of the adrenal medulla and sympathetic ganglia.
- The pathophysiology of pheochromocytoma does not depend on the histological subtype. Malignant and benign pheochromocytomas share the same biochemical and histological features. [2][3][4]
- The exact mechanism responsible for surge in catecholamine secretion remains unclear but it has been postulated that certain medications (such as opiates, metoclopramide or beta blockers) and changes in tumor blood flow and pressure could be responsible factors.
- Binding to β1 receptors causes renin release from juxtaglomerular cells and lipolysis in adipose tissue. It Increases cardiac output by:
- Increase in heart rate in sinoatrial node
- Increase in atrial cardiac muscle contractility
- Increases in contractility and automaticity of ventricular cardiac muscle
- Increases in conduction and automaticity of atrioventricular node
Genetics
- Genes involved in the pathogenesis of pheochromocytoma include:
- RET gene (MEN 2A, MEN 2B syndromes)
- NF1 gene
- VHL gene (VHL disease)
- SDHD, SDHB, and SDHC genes of the mitochondrial complex [7]
- SDHA, SDHAF2, TMEM127 (transmembrane protein 127), MAX (myc-associated factor X), FH (fumarate hydratase), PDH1, PDH2 (pyruvate dehydrogenase), HIF1alpha (hypoxia-inducible factor), MDH2 (malate dehydrogenase), and KIF1Bß (kinesin family member) genes. [8]
Pheochromocytoma and paragangliomas (PPGL) susceptibility genes can be classified into the following clusters- [9] [10] [11]
- Cluster 1
- Mutations involving in overexpression of vascular endothelial growth factor (VEGF) as a result of pseudohypoxia
- Impaired DNA methylation leading to increased vascularization
- Cluster 2
- Activating mutations of Wnt-signaling pathway including Wnt receptor signaling and Hedgehog signaling.
- Mutations of CSDE1 (Cold shock domain containing E1) and MAML3 (Mastermind like transcriptional coactivator 3) genes7.
- Cluster 3
- Abnormal activation of kinase signaling pathways like PI3Kinase/AKT, RAS/RAF/ERK, and mTOR pathways.
Associated conditions
Conditions associated with pheochromocytoma include:
- Multiple endocrine neoplasia (MEN1)
- Multiple endocrine neoplasia (MEN2B)
- Von-Hippel Lindau disease (VHL)
- Neurofibromatosis 1 (NF1)
| MEN 1 | MEN 2 |
|---|---|
Gross Pathology
On gross pathology, the characteristic findings of pheochromocytoma are:
- Small to large tumors usually associated with hemorrhage and necrosis.[12]
- Usually lobulated
- Bilateral when familial tumors
- Associated with hyperplasia in the adjacent medulla.
- Chromaffin reaction: fresh tumor cut section turns dark brown if add potassium dichromate at pH 5-6.
-
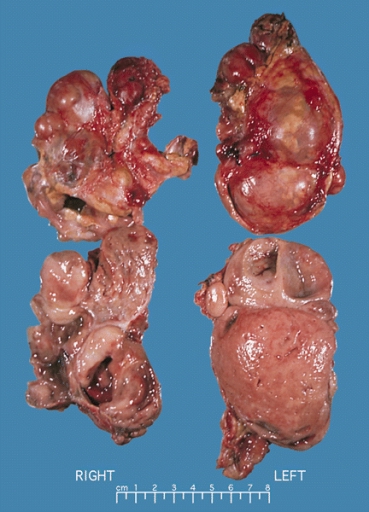 Bilateral pheochromocytoma in MEN2. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg
Bilateral pheochromocytoma in MEN2. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg

Microscopic Pathology
On microscopic histopathological analysis, the characterisitc findings of pheochromocytoma typically include: [13] [14]
- A nesting (Zellballen) pattern- this pattern is composed of well-defined clusters of tumor cells (round or polygonal epithelioid cells) containing eosinophilic cytoplasm separated by fibrovascular stroma.
- These cells have a central nucleus with an eosinophilic, granular cytoplasm, and clumped chromatin.
- At the periphery, spindle-shaped sustentacular or supporting cells are seen.
-
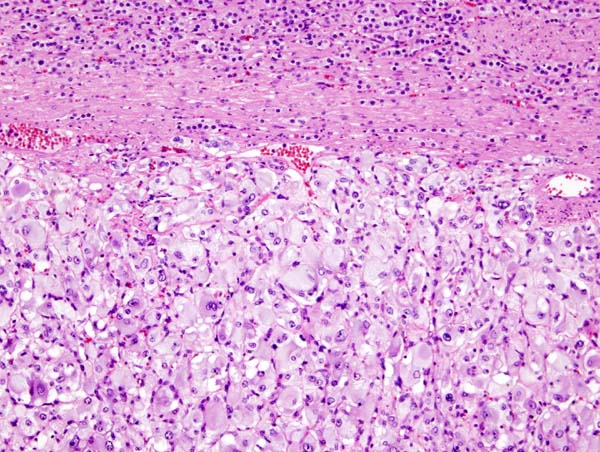 Micrograph of pheochromocytoma. Source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524
Micrograph of pheochromocytoma. Source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524 -
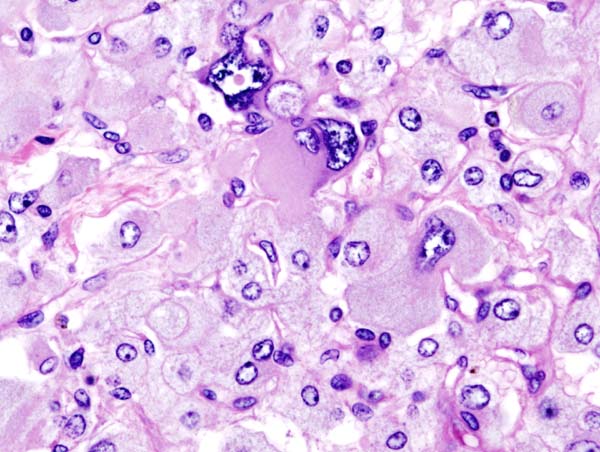 Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945 -
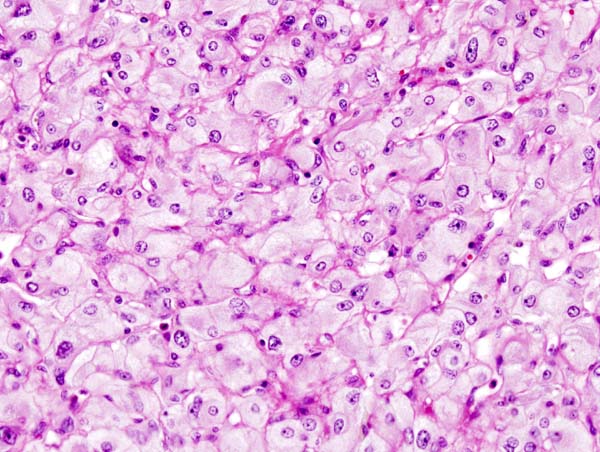 Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944
Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944
_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
{kind=link}
Videos
{{#ev:youtube|7yjxG3KmX98}}
References
- ↑ Smith RJ, Bryant RG (1975). "Metal substitutions incarbonic anhydrase: a halide ion probe study". Biochem Biophys Res Commun. 66 (4): 1281–6. doi:10.1016/0006-291x(75)90498-2. PMID orcid.org/0000-0003-2771-564X Check
|pmid=value (help). - ↑ Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA; et al. (1999). "Clinical experience over 48 years with pheochromocytoma". Ann Surg. 229 (6): 755–64, discussion 764-6. PMC 1420821. PMID 10363888.
- ↑ Raz I, Katz A, Spencer MK (1991). "Epinephrine inhibits insulin-mediated glycogenesis but enhances glycolysis in human skeletal muscle". Am J Physiol. 260 (3 Pt 1): E430–5. PMID 1900669.
- ↑ Arnall DA, Marker JC, Conlee RK, Winder WW (1986). "Effect of infusing epinephrine on liver and muscle glycogenolysis during exercise in rats". Am J Physiol. 250 (6 Pt 1): E641–9. PMID 3521311.
- ↑ Webb TA, Sheps SG, Carney JA (1980). "Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2". Am. J. Surg. Pathol. 4 (2): 121–6. PMID 6103678.
- ↑ Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T (1987). "Gene expression from transcriptionally disabled retroviral vectors". Proc. Natl. Acad. Sci. U.S.A. 84 (15): 5197–201. PMC 298821. PMID 3474647.
- ↑ Gimm O (2005). "Pheochromocytoma-associated syndromes: genes, proteins and functions of RET, VHL and SDHx". Fam Cancer. 4 (1): 17–23. doi:10.1007/s10689-004-5740-1. PMID 15883706.
- ↑ Jameson, J (2017). Harrison's Principles of Internal Medicine 19th Edition and Harrison's Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Jameson, J (2017). Harrison's Principles of Internal Medicine 19th Edition and Harrison's Manual of Medicine 19th Edition VAL PAK. New York: McGraw-Hill Medical. ISBN 978-1260128857.
- ↑ Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM; et al. (2004). "Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome". Endocr Relat Cancer. 11 (4): 897–911. doi:10.1677/erc.1.00838. PMID 15613462.
- ↑ Lam AK (2017). "Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours". Endocr Pathol. 28 (3): 213–227. doi:10.1007/s12022-017-9484-5. PMID 28477311.
- ↑ Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB (2015). "Non Functional Unilateral Adrenal Myelolipoma, A Case Report". J Clin Diagn Res. 9 (6): ED03–4. doi:10.7860/JCDR/2015/13209.6070. PMC 4525519. PMID 26266130.
- ↑ Kliewer KE, Wen DR, Cancilla PA, Cochran AJ (1989). "Paragangliomas: assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniques". Hum Pathol. 20 (1): 29–39. doi:10.1016/0046-8177(89)90199-8. PMID 2912871.
- ↑ Kliewer KE, Cochran AJ (1989). "A review of the histology, ultrastructure, immunohistology, and molecular biology of extra-adrenal paragangliomas". Arch Pathol Lab Med. 113 (11): 1209–18. PMID 2684087.