Valdecoxib
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Rabin Bista, M.B.B.S. [2]
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
Serious Skin Reactions:
See full prescribing information for complete Boxed Warning.
Serious Skin Reactions:
|
Overview
Valdecoxib is a Cyclooxygenase-2 Inhibitor that is FDA approved for the treatment of osteoarthritis, adult rheumatoid arthritis and primary dysmenorrhea. There is a Black Box Warning for this drug as shown here. Common adverse reactions include Abdominal pain, Diarrhea, Flatulence, Indigestion, Nausea, Dizziness, Headache, Rash, Hypertension, Peripheral edema , Backache, Myalgia.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Indications
- For relief of the signs and symptoms of osteoarthritis and adult rheumatoid arthritis.
- For the treatment of primary dysmenorrhea
Dosage
Osteoarthritis and Adult Rheumatoid Arthritis
- The recommended dose of BEXTRA Tablets for the relief of the signs and symptoms of arthritis is 10 mg once daily.
Primary Dysmenorrhea
- The recommended dose of BEXTRA Tablets for treatment of primary dysmenorrhea is 20 mg twice daily, as needed.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Valdecoxib in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Valdecoxib in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Valdecoxib in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Valdecoxib in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Valdecoxib in pediatric patients.
Contraindications
- BEXTRA should not be given to patients who have demonstrated allergic-type reactions to sulfonamides.
- BEXTRA Tablets are contraindicated in patients with known hypersensitivity to valdecoxib. BEXTRA should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs are possible in such patients.
- BEXTRA is contraindicated for the treatment of post-operative pain immediately following coronary artery bypass graft (CABG) surgery and should not be used in this setting.
Warnings
|
Serious Skin Reactions:
See full prescribing information for complete Boxed Warning.
Serious Skin Reactions:
|
Gastrointestinal (GI) Effects — Risk of GI Ulceration, Bleeding, and Perforation
- Serious gastrointestinal toxicity such as bleeding, ulceration and perforation of the stomach, small intestine or large intestine can occur at any time with or without warning symptoms in patients treated with nonsteroidal anti-inflammatory drugs (NSAIDs). Minor gastrointestinal problems such as dyspepsia are common and may also occur at any time during NSAID therapy. Therefore, physicians and patients should remain alert for ulceration and bleeding even in the absence of previous GI tract symptoms. Patients should be informed about the signs and symptoms of serious GI toxicity and the steps to take if they occur. The utility of periodic laboratory monitoring has not been demonstrated, nor has it been adequately assessed. Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. It has been demonstrated that upper GI ulcers, gross bleeding or perforation caused by NSAIDs appear to occur in approximately 1% of patients treated for 3 to 6 months and 2–4% of patients treated for one year. These trends continue, thus increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk.
- NSAIDs should be prescribed with extreme caution in patients with a prior history of ulcer disease or gastrointestinal bleeding. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore special care should be taken in treating this population. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
- Studies have shown that patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding and who use NSAIDs, have a greater than 10-fold higher risk for developing a GI bleed than patients with neither of these risk factors. In addition to a past history of ulcer disease, pharmacoepidemiological studies have identified several other co-therapies or co-morbid conditions that may increase the risk for GI bleeding such as: treatment with oral corticosteroids, treatment with anticoagulants, longer duration of NSAID therapy, smoking, alcoholism, older age, and poor general health status.
Serious Skin Reactions
- Valdecoxib contains a sulfonamide moiety and patients with a known history of a sulfonamide allergy may be at a greater risk of skin reactions. Patients without a history of sulfonamide allergy may also be at risk for serious skin reactions.
- Serious skin reactions, including erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis, have been reported through postmarketing surveillance in patients receiving BEXTRA . Fatalities due to Stevens-Johnson syndrome and toxic epidermal necrolysis have been reported. Patients appear to be at higher risk for these events early in the course of therapy, with the onset of the event occurring in the majority of cases within the first two weeks of treatment. BEXTRA should be discontinued at the first appearance of skin rash, mucosal lesions or any other sign of hypersensitivity. Serious skin reactions have been reported with other COX-2 inhibitors during postmarketing experience. The reported rate of these events appears to be greater for BEXTRA as compared to other COX-2 agents.
Anaphylactoid Reactions
- In postmarketing experience, cases of hypersensitivity reactions (anaphylactic reactions and angioedema) have been reported in patients receiving BEXTRA. These cases have occurred in patients with and without a history of allergic-type reactions to sulfonamides. BEXTRA should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs.
- Emergency help should be sought in cases where an anaphylactoid reaction occurs.
Coronary Artery Bypass Graft Surgery
- Patients treated with BEXTRA for pain following coronary artery bypass graft surgery have a higher risk for cardiovascular/thromboembolic events, deep surgical infections or sternal wound complications. BEXTRA is therefore contraindicated for the treatment of postoperative pain following CABG surgery.
Advanced Renal Disease
- No information is available regarding the safe use of BEXTRA Tablets in patients with advanced kidney disease. Therefore, treatment with BEXTRA is not recommended in these patients. If therapy with BEXTRA must be initiated, close monitoring of the patient's kidney function is advisable.
Pregnancy
- In late pregnancy, BEXTRA should be avoided because it may cause premature closure of the ductus arteriosus.
Precautions
General
- BEXTRA Tablets cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to exacerbation of corticosteroid-responsive illness. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
- The pharmacological activity of valdecoxib in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Hepatic Effects
- Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials with NSAIDs. These laboratory abnormalities may progress, may remain unchanged, or may remain transient with continuing therapy. Rare cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, liver necrosis and hepatic failure (some with fatal outcome) have been reported with NSAIDs. In controlled clinical trials of valdecoxib, the incidence of borderline (defined as 1.2- to 3.0-fold) elevations of liver tests was 8.0% for valdecoxib and 8.4% for placebo, while approximately 0.3% of patients taking valdecoxib, and 0.2% of patients taking placebo, had notable (defined as greater than 3-fold) elevations of ALT or AST.
- A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be monitored carefully for evidence of the development of a more severe hepatic reaction while on therapy with BEXTRA. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash), BEXTRA should be discontinued.
Renal Effects
- Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a nonsteroidal anti-inflammatory drug may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and Angiotensin Converting Enzyme (ACE) inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
- Caution should be used when initiating treatment with BEXTRA in patients with considerable dehydration. It is advisable to rehydrate patients first and then start therapy with BEXTRA. Caution is also recommended in patients with preexisting kidney disease.
Hematological Effects
- Anemia is sometimes seen in patients receiving BEXTRA. Patients on long-term treatment with BEXTRA should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
- BEXTRA does not generally affect platelet counts, prothrombin time (PT), or activated partial thromboplastin time (APTT), and does not appear to inhibit platelet aggregation at indicated dosages.
Fluid Retention and Edema
- Fluid retention and edema have been observed in some patients taking BEXTRA. Therefore, BEXTRA should be used with caution in patients with fluid retention, hypertension, or heart failure.
Preexisting Asthma
- Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm, which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and other nonsteroidal anti-inflammatory drugs has been reported in such aspirin-sensitive patients, BEXTRA should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Laboratory Tests
- Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs and symptoms of GI bleeding.
Adverse Reactions
Clinical Trials Experience
- Of the patients treated with BEXTRA Tablets in controlled arthritis trials, 2665 were patients with OA, and 2684 were patients with RA. More than 4000 patients have received a chronic total daily dose of BEXTRA 10 mg or more. More than 2800 patients have received BEXTRA 10 mg/day, or more, for at least 6 months and 988 of these have received BEXTRA for at least 1 year.
Osteoarthritis and Rheumatoid Arthritis
- Table 4 lists all adverse events, regardless of causality, that occurred in ≥2.0% of patients receiving BEXTRA 10 and 20 mg/day in studies of three months or longer from 7 controlled studies conducted in patients with OA or RA that included a placebo and/or a positive control group.

- In these placebo- and active-controlled clinical trials, the discontinuation rate due to adverse events was 7.5% for arthritis patients receiving valdecoxib 10 mg daily, 7.9% for arthritis patients receiving valdecoxib 20 mg daily and 6.0%for patients receiving placebo.
- In the seven controlled OA and RA studies, the following adverse events occurred in 0.1–1.9% of patients treated with BEXTRA 10–20 mg daily, regardless of causality.
Application site disorders
Cardiovascular
- Aggravated hypertension, aneurysm, angina pectoris, arrhythmia, cardiomyopathy, congestive heart failure, coronary artery disorder, heart murmur, hypotension
Central, peripheral nervous system
- Cerebrovascular disorder, hypertonia, hypoesthesia, migraine, neuralgia, neuropathy, paresthesia, tremor, twitching, vertigo
Endocrine
Female reproductive
- Amenorrhea, dysmenorrhea, leukorrhea, mastitis, menstrual disorder, menorrhagia, menstrual bloating, vaginal hemorrhage
Gastrointestinal
- Abnormal stools, constipation, diverticulosis, dry mouth, duodenal ulcer, duodenitis, eructation, esophagitis, fecal incontinence, gastric ulcer, gastritis, gastroenteritis, gastroesophageal reflux, hematemesis, hematochezia, hemorrhoids, hemorrhoids bleeding, hiatal hernia, melena, stomatitis, stool frequency increased, tenesmus, tooth disorder, vomiting
General
- Allergy aggravated, allergic reaction, asthenia, chest pain, chills, cyst NOS, generalized edema, face edema, fatigue, fever, hot flushes, halitosis, malaise, pain, periorbital swelling, peripheral pain
Hearing and vestibular
- Ear abnormality, earache, tinnitus
Heart rate and rhythm
Hemic
Liver and biliary system
Male reproductive
- Impotence, prostatic disorder
Metabolic and nutritional
- Alkaline phosphatase increased, BUN increased, CPK increased, creatinine increased, diabetes mellitus, glycosuria, gout, hypercholesterolemia, hyperglycemia, hyperkalemia, hyperlipemia, hyperuricemia, hypocalcemia, hypokalemia, LDH increased, thirst increased, weight loss, weight gain, xerophthalmia
Musculoskeletal
- Arthralgia, fracture accidental, neck stiffness, osteoporosis, synovitis, tendonitis
Neoplasm
- Breast neoplasm, lipoma, malignant ovarian cyst
Platelets (bleeding or clotting)
Psychiatric
- Anorexia, anxiety, appetite increased, confusion, depression, depression aggravated, insomnia, nervousness, morbid dreaming, somnolence
Resistance mechanism disorders
- Herpes simplex, herpes zoster, fungal infection, soft tissue infection, viral infection, moniliasis, moniliasis genital, otitis media
Respiratory
- Abnormal breath sounds, bronchitis, bronchospasm, coughing, dyspnea, emphysema, laryngitis, pneumonia, pharyngitis, pleurisy, rhinitis
Skin and appendages
- Acne, alopecia, dermatitis, fungal dermatitis , eczema, photosensitivity allergic reaction, pruritus, erythematous rash, maculopapular rash, psoriaform rash, skin dry, skin hypertrophy, skin ulceration, increased sweating, urticaria
Special senses
Urinary system
- Albuminuria, cystitis, dysuria, hematuria, micturition frequency increased, pyuria, urinary incontinence, urinary tract infection
Vascular
Vision
- Blurred vision, cataract, conjunctival hemorrhage, conjunctivitis, eye pain, keratitis, abnormal vision
White cell and RES disorders
- Other serious adverse events that were reported rarely (estimated <0.1%) in clinical trials, regardless of causality, in patients taking BEXTRA:
Autonomic nervous system disorders
- Hypertensive encephalopathy, vasospasm
Cardiovascular
- Abnormal ECG, aortic stenosis, atrial fibrillation, carotid stenosis, coronary thrombosis, heart block, heart valve disorders, mitral insufficiency, myocardial infarction, myocardial ischemia, pericarditis, syncope, thrombophlebitis, unstable angina, ventricular fibrillation
Central, peripheral nervous system
Endocrine
Female reproductive
Gastrointestinal
- Appendicitis, colitis with bleeding, dysphagia, esophageal perforation, gastrointestinal bleeding, ileus, intestinal obstruction, peritonitis
Hemic
Liver and biliary system
Metabolic
Musculoskeletal
Neoplasm
- Benign brain neoplasm, bladder carcinoma, carcinoma, gastric carcinoma, prostate carcinoma, pulmonary carcinoma
Platelets (bleeding or clotting)
Psychiatric
Renal
Resistance mechanism disorders
- Sepsis
Respirator
- Apnea, pleural effusion, pulmonary edema, pulmonary fibrosis, pulmonary infarction, pulmonary hemorrhage, respiratory insufficiency
Skin
Urinary system
Vision
Postmarketing Experience
- The following reactions have been identified during postmarketing use of BEXTRA. These reactions have been chosen for inclusion either due to their seriousness, reporting frequency, possible causal relationship to BEXTRA, or a combination of these factors. Because these reactions were reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- General: Hypersensitivity reactions (including anaphylactic reactions and angioedema)
- Gastrointestinal: Pancreatitis
- Skin and appendages: Erythema multiforme, exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis
Drug Interactions
- The drug interaction studies with valdecoxib were performed both with valdecoxib and a rapidly hydrolyzed intravenous prodrug form. The results from trials using the intravenous prodrug are reported in this section as they relate to the role of valdecoxib in drug interactions.
General
- In humans, valdecoxib metabolism is predominantly mediated via CYP 3A4 and 2C9 with glucuronidation being a further (20%) route of metabolism. In vitro studies indicate that valdecoxib is a moderate inhibitor of CYP 2C19 (IC50 = 6 µg/mL or 19 µM) and 2C9 (IC50 = 13 µg/mL or 41 µM), and a weak inhibitor of CYP 2D6 (IC50 = 31 µg/mL or 100 µM) and 3A4 (IC50 = 44 µg/mL or 141 µM).
Aspirin
- Concomitant administration of aspirin with valdecoxib may result in an increased risk of GI ulceration and complications compared to valdecoxib alone. Because of its lack of anti-platelet effect valdecoxib is not a substitute for aspirin for cardiovascular prophylaxis.
- In a parallel group drug interaction study comparing the intravenous prodrug form of valdecoxib at 40 mg BID (n=10) vs placebo (n=9), valdecoxib had no effect on in vitro aspirin-mediated inhibition of arachidonate- or collagen-stimulated platelet aggregation.
Methotrexate
- Valdecoxib 10 mg BID did not show a significant effect on the plasma exposure or renal clearance of methotrexate.
ACE-inhibitors
- Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking BEXTRA concomitantly with ACE-inhibitors.
Furosemide
- Clinical studies, as well as post-marketing observations, have shown that NSAIDs can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis.
Anticonvulsants (Phenytoin)
- Steady state plasma exposure (AUC) of valdecoxib (40 mg BID for 12 days) was decreased by 27% when coadministered with multiple doses (300 mg QD for 12 days) of phenytoin (a CYP 3A4 inducer). Patients already stabilized on valdecoxib should be closely monitored for loss of symptom control with phenytoin coadministration. Valdecoxib did not have a statistically significant effect on the pharmacokinetics of phenytoin (a CYP 2C9 and CYP 2C19 substrate).
- Drug interaction studies with other anticonvulsants have not been conducted. Routine monitoring should be performed when therapy with BEXTRA is either initiated or discontinued in patients on anticonvulsant therapy.
Dextromethorphan
- Dextromethorphan is primarily metabolized by CYP 2D6 and to a lesser extent by 3A4. Coadministration with valdecoxib (40 mg BID for 7 days) resulted in a significant increase in dextromethorphan plasma levels suggesting that, at these doses, valdecoxib is a weak inhibitor of 2D6. Even so, dextromethorphan plasma concentrations in the presence of high doses of valdecoxib were almost 5-fold lower than those seen in CYP 2D6 poor metabolizers suggesting that dose adjustment is not necessary.
Lithium
- Valdecoxib 40 mg BID for 7 days produced significant decreases in lithium serum clearance (25%) and renal clearance (30%) with a 34% higher serum exposure compared to lithium alone. Lithium serum concentrations should be monitored closely when initiating or changing therapy with BEXTRA in patients receiving lithium. Lithium carbonate (450 mg BID for 7 days) had no effect on valdecoxib pharmacokinetics.
Warfarin
- The effect of valdecoxib on the anticoagulant effect of warfarin (1–8 mg/day) was studied in healthy subjects by coadministration of BEXTRA 40 mg BID for 7 days. Valdecoxib caused a statistically significant increase in plasma exposures of R-warfarin and S-warfarin (12% and 15%, respectively), and in the pharmacodynamic effects (prothrombin time, measured as INR) of warfarin. While mean INR values were only slightly increased with coadministration of valdecoxib, the day-to-day variability in individual INR values was increased. Anticoagulant therapy should be monitored, particularly during the first few weeks, after initiating therapy with BEXTRA in patients receiving warfarin or similar agents.
Fluconazole and Ketoconazole
- Ketoconazole and fluconazole are predominantly CYP 3A4 and 2C9 inhibitors, respectively. Concomitant single dose administration of valdecoxib 20 mg with multiple doses of ketoconazole and fluconazole produced a significant increase in exposure of valdecoxib. Plasma exposure (AUC) to valdecoxib was increased 62% when coadministered with fluconazole and 38% when coadministered with ketoconazole.
Glyburide
- Glyburide is a CYP 2C9 substrate. Coadministration of valdecoxib (10 mg BID for 7 days) with glyburide (5 mg QD or 10 mg BID) did not affect the pharmacokinetics (exposure) of glyburide. Coadministration of valdecoxib (40 mg BID (day 1) and 40 mg QD (days 2–7)) with glyburide (5 mg QD) did not affect either the pharmacokinetics (exposure) or the pharmacodynamics (blood glucose and insulin levels) of glyburide. Coadministration of valdecoxib (40 mg BID (day 1) and 40 mg QD (days 2–7)) with glyburide (10 mg glyburide BID) resulted in 21% increase in glyburide AUC(0–12hr) and a 16% increase in glyburide Cmax leading to a 16%decrease in glucose AUC(0–24hr). Insulin parameters were not affected. Because changes in glucose concentrations with valdecoxib coadministration were within the normal variability and individual glucose concentrations were above or near 70 mg/dL, dose adjustment for glyburide (5 mg QD and 10 mg BID) with valdecoxib coadministration (up to 40 mg QD) is not indicated. Coadministration of glyburide with doses higher than 40 mg valdecoxib (e.g., 40 mg BID) has not been studied.
Omeprazole
- Omeprazole is a CYP 3A4 substrate and CYP 2C19 substrate and inhibitor. Valdecoxib steady state plasma concentrations (40 mg BID) were not affected significantly with multiple doses of omeprazole (40 mg QD). Coadministration with valdecoxib increased exposure of omeprazole (AUC) by 46%. Drugs whose absorption is sensitive to pH may be negatively impacted by concomitant administration of omeprazole and valdecoxib. However, because higher doses (up to 360 mg QD) of omeprazole are tolerated in Zollinger-Ellison (ZE) patients, no dose adjustment for omeprazole is recommended at current doses. Coadministration of valdecoxib with doses higher than 40 mg QD omeprazole has not been studied.
Oral Contraceptives
- Valdecoxib (40 mg BID) did not induce the metabolism of the combination oral contraceptive norethindrone/ethinyl estradiol (1 mg/0.035 mg combination, Ortho-Novum 1/35®). Coadministration of valdecoxib and Ortho-Novum 1/35® increased the exposure of norethindrone and ethinyl estradiol by 20% and 34%, respectively. Although there is little risk for loss of contraceptive efficacy, the clinical significance of these increased exposures in terms of safety is not known. These increased exposures of norethindrone and ethinyl estradiol should be taken into consideration when selecting an oral contraceptive for women taking valdecoxib.
Diazepam
- Diazepam (Valium®) is a CYP 3A4 and CYP 2C19 substrate. Plasma exposure of diazepam (10 mg BID) was increased by 28% following administration of valdecoxib (40 mg BID) for 12 days, while plasma exposure of valdecoxib (40 mg BID) was not substantially increased following administration of diazepam (10 mg BID) for 12 days. Although the magnitude of changes in diazepam plasma exposure when coadministered with valdecoxib were not sufficient to warrant dosage adjustments, patients may experience enhanced sedative side effects caused by increased exposure of diazepam under this circumstance. Patients should be cautioned against engaging in hazardous activities requiring complete mental alertness such as operating machinery or driving a motor vehicle.
Use in Specific Populations
Pregnancy
Teratogenic Effects
- The incidence of fetuses with skeletal anomalies such as semi-bipartite thoracic vertebra centra and fused sternebrae was slightly higher in rabbits at an oral dose of 40 mg/kg/day (equivalent to approximately 72-fold human exposures at 20 mg QD as measured by the AUC(0–24hr)) throughout organogenesis. Valdecoxib was not teratogenic in rabbits up to an oral dose of 10 mg/kg/day (equivalent to approximately 8-fold human exposures at 20 mg QD as measured by the AUC(0–24hr)).
- Valdecoxib was not teratogenic in rats up to an oral dose of 10 mg/kg/day (equivalent to approximately 19-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)). There are no studies in pregnant women. However, valdecoxib crosses the placenta in rats and rabbits. BEXTRA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Non-teratogenic Effects
- Valdecoxib caused increased pre- and post-implantation loss with reduced live fetuses at oral doses ≥10 mg/kg/day (equivalent to approximately 19-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)) in rats and an oral dose of 40 mg/kg/day (equivalent to approximately 72-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)) in rabbits throughout organogenesis. In addition, reduced neonatal survival and decreased neonatal body weight when rats were treated with valdecoxib at oral doses ≥6 mg/kg/day (equivalent to approximately 7-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)) throughout organogenesis and lactation period. No studies have been conducted to evaluate the effect of valdecoxib on the closure of the ductus arteriosus in humans. Therefore, as with other drugs known to inhibit prostaglandin synthesis, use of BEXTRA during the third trimester of pregnancy should be avoided.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Valdecoxib in women who are pregnant.
Labor and Delivery
- Valdecoxib produced no evidence of delayed labor or parturition at oral doses up to 10 mg/kg/day in rats (equivalent to approximately 19-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)). The effects of BEXTRA on labor and delivery in pregnant women are unknown.
Nursing Mothers
- Valdecoxib and its active metabolite are excreted in the milk of lactating rats. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for adverse reactions in nursing infants from BEXTRA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother and the importance of nursing to the infant.
Pediatric Use
- Safety and effectiveness of BEXTRA in pediatric patients below the age of 18 years have not been evaluated.
Geriatic Use
- Of the patients who received BEXTRA in arthritis clinical trials of three months duration, or greater, approximately 2100 were 65 years of age or older, including 570 patients who were 75 years or older. No overall differences in effectiveness were observed between these patients and younger patients.
Gender
There is no FDA guidance on the use of Valdecoxib with respect to specific gender populations.
Race
There is no FDA guidance on the use of Valdecoxib with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Valdecoxib in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Valdecoxib in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Valdecoxib in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Valdecoxib in patients who are immunocompromised.
Administration and Monitoring
Administration
- Oral
Monitoring
- If therapy with BEXTRA must be initiated, close monitoring of the patient's kidney function is advisable
- A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be monitored carefully for evidence of the development of a more severe hepatic reaction while on therapy with BEXTRA.
- Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs and symptoms of GI bleeding.
- Patients already stabilized on valdecoxib should be closely monitored for loss of symptom control with phenytoin coadministration.
- Lithium serum concentrations should be monitored closely when initiating or changing therapy with BEXTRA in patients receiving lithium.
- Anticoagulant therapy should be monitored, particularly during the first few weeks, after initiating therapy with BEXTRA in patients receiving warfarin or similar agents.
IV Compatibility
There is limited information regarding IV Compatibility of Valdecoxib in the drug label.
Overdosage
- Symptoms following acute NSAID overdoses are usually limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which are generally reversible with supportive care. Gastrointestinal bleeding can occur. Hypertension, acute renal failure, respiratory depression and coma may occur, but are rare.
- Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose.
- Patients should be managed by symptomatic and supportive care following an NSAID overdose. There are no specific antidotes. Hemodialysis removed only about 2% of administered valdecoxib from the systemic circulation of 8 patients with end-stage renal disease and, based on its degree of plasma protein binding (>98%), dialysis is unlikely to be useful in overdose. Forced diuresis, alkalinization of urine, or hemoperfusion also may not be useful due to high protein binding.
Pharmacology


Mechanism of Action
- Valdecoxib is a nonsteroidal anti-inflammatory drug (NSAID) that exhibits anti-inflammatory, analgesic and antipyretic properties in animal models. The mechanism of action is believed to be due to inhibition of prostaglandin synthesis primarily through inhibition of cyclooxygenase-2 (COX-2). At therapeutic plasma concentrations in humans valdecoxib does not inhibit cyclooxygenase-1 (COX-1).
Structure
- Valdecoxib is chemically designated as 4-(5-methyl-3-phenyl-4-isoxazolyl) benzenesulfonamide and is a diaryl substituted isoxazole. It has the following chemical structure:
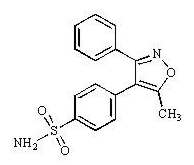
This image is provided by the National Library of Medicine.

- The empirical formula for valdecoxib is C16H14N2O3S, and the molecular weight is 314.36. Valdecoxib is a white crystalline powder that is relatively insoluble in water (10 µg/mL) at 25°C and pH 7.0, soluble in methanol and ethanol, and freely soluble in organic solvents and alkaline (pH=12) aqueous solutions.
- BEXTRA Tablets for oral administration contain either 10 mg or 20 mg of valdecoxib. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, pregelatinized starch, croscarmellose sodium, magnesium stearate, hypromellose, polyethylene glycol, polysorbate 80, and titanium dioxide.
Pharmacodynamics
There is limited information regarding Pharmacodynamics of Valdecoxib in the drug label.
Pharmacokinetics
Absorption
- Valdecoxib achieves maximal plasma concentrations in approximately 3 hours. The absolute bioavailability of valdecoxib is 83% following oral administration of BEXTRA compared to intravenous infusion of valdecoxib.
- Dose proportionality was demonstrated after single doses (1–400 mg) of valdecoxib. With multiple doses (up to 100 mg/day for 14 days), valdecoxib exposure as measured by the AUC, increases in a more than proportional manner at doses above 10 mg BID. Steady state plasma concentrations of valdecoxib are achieved by day 4.
- The steady state pharmacokinetic parameters of valdecoxib in healthy male subjects are shown in Table 1.
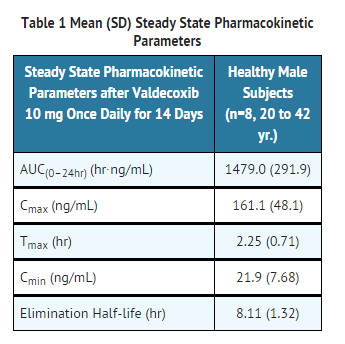
This image is provided by the National Library of Medicine.

- No clinically significant age or gender differences were seen in pharmacokinetic parameters that would require dosage adjustments.
- Effect of Food and Antacid
- BEXTRA can be taken with or without food. Food had no significant effect on either the peak plasma concentration (Cmax) or extent of absorption (AUC) of valdecoxib when BEXTRA was taken with a high fat meal. The time to peak plasma concentration (Tmax), however, was delayed by 1–2 hours. Administration of BEXTRA with antacid (aluminum/magnesium hydroxide) had no significant effect on either the rate or extent of absorption of valdecoxib.
Distribution
- Plasma protein binding for valdecoxib is about 98% over the concentration range (21–2384 ng/mL). Steady state apparent volume of distribution (Vss/F) of valdecoxib is approximately 86 L after oral administration. Valdecoxib and its active metabolite preferentially partition into erythrocytes with a blood to plasma concentration ratio of about 2.5:1. This ratio remains approximately constant with time and therapeutic blood concentrations.
Metabolism
- In humans, valdecoxib undergoes extensive hepatic metabolism involving both P450 isoenzymes (3A4 and 2C9) and non-P450 dependent pathways (i.e., glucuronidation). Concomitant administration of BEXTRA with known CYP 3A4 and 2C9 inhibitors (e.g., fluconazole and ketoconazole) can result in increased plasma exposure of valdecoxib.
- One active metabolite of valdecoxib has been identified in human plasma at approximately 10% the concentration of valdecoxib. This metabolite, which is a less potent COX-2 specific inhibitor than the parent, also undergoes extensive metabolism and constitutes less than 2% of the valdecoxib dose excreted in the urine and feces. Due to its low concentration in the systemic circulation, it is not likely to contribute significantly to the efficacy profile of BEXTRA.
Excretion
- Valdecoxib is eliminated predominantly via hepatic metabolism with less than 5% of the dose excreted unchanged in the urine and feces. About 70% of the dose is excreted in the urine as metabolites, and about 20% as valdecoxib N-glucuronide. The apparent oral clearance (CL/F) of valdecoxib is about 6 L/hr. The mean elimination half-life (T1/2) ranges from 8–11 hours, and increases with age.
Special Populations
Geriatric
- In elderly subjects (> 65 years), weight-adjusted steady state plasma concentrations (AUC(0–12hr)) are about 30% higher than in young subjects. No dose adjustment is needed based on age.
Pediatric
- BEXTRA has not been investigated in pediatric patients below 18 years of age.
Race
- Pharmacokinetic differences due to race have not been identified in clinical and pharmacokinetic studies conducted to date.
Hepatic Insufficiency
- Valdecoxib plasma concentrations are significantly increased (130%) in patients with moderate (Child-Pugh Class B) hepatic impairment. In clinical trials, doses of BEXTRA above those recommended have been associated with fluid retention. Hence, treatment with BEXTRA should be initiated with caution in patients with mild to moderate hepatic impairment and fluid retention. The use of BEXTRA in patients with severe hepatic impairment (Child-Pugh Class C) is not recommended.
Renal Insufficiency
- The pharmacokinetics of valdecoxib have been studied in patients with varying degrees of renal impairment. Because renal elimination of valdecoxib is not important to its disposition, no clinically significant changes in valdecoxib clearance were found even in patients with severe renal impairment or in patients undergoing renal dialysis. In patients undergoing hemodialysis the plasma clearance (CL/F) of valdecoxib was similar to the CL/F found in healthy elderly subjects (CL/F about 6 to 7 L/hr.) with normal renal function (based on creatinine clearance).
- NSAIDs have been associated with worsening renal function and use in advanced renal disease is not recommended (see PRECAUTIONS — RENAL EFFECTS).
Drug Interactions
- For quantitative information on the following drug interaction studies, see PRECAUTIONS — DRUG INTERACTIONS.
General
- Valdecoxib undergoes both P450 (CYP) dependent and non-P450 dependent (glucuronidation) metabolism. In vitro studies indicate that valdecoxib is not a significant inhibitor of CYP 1A2, 3A4, or 2D6 and is a weak inhibitor of CYP 2C9 and a weak to moderate inhibitor of CYP 2C19 at therapeutic concentrations. The P450-mediated metabolic pathway of valdecoxib predominantly involves the 3A4 and 2C9 isozymes. Using prototype inhibitors and substrates of these isozymes, the following results were obtained. Coadministration of a known inhibitor of CYP 2C9/3A4 (fluconazole) and a CYP 3A4 inhibitor (ketoconazole) enhanced the total plasma exposure (AUC) of valdecoxib. Coadministration of valdecoxib with a CYP 3A4 inducer (phenytoin) decreased total plasma exposure (AUC) of valdecoxib.
- Coadministration of valdecoxib with warfarin (a CYP 2C9 substrate) caused a small, but statistically significant increase in plasma exposures of R-warfarin and S-warfarin, and also in the pharmacodynamic effects (International Normalized Ratio-INR) of warfarin.
- Coadministration of valdecoxib with diazepam (a CYP 2C19/3A4 substrate) resulted in increased exposure of diazepam, but not its major metabolite, desmethyldiazepam.
- Coadministration of valdecoxib with glyburide (a CYP 2C9 substrate) (40 mg valdecoxib QD with 10 mg glyburide BID) resulted in increased exposure of glyburide.
- Coadministration of valdecoxib with an oral contraceptive, 1 mg norethindrone/0.035 mg ethinyl estradiol (CYP 3A4 substrates), resulted in increased exposure of both norethindrone and ethinyl estradiol.
- Coadministration of valdecoxib with omeprazole (a CYP 3A4/2C19 substrate) caused an increase in omeprazole exposure.
- Coadministration of valdecoxib with dextromethorphan (a CYP 2D6/3A4 substrate) resulted in an increase in dextromethorphan plasma levels above those seen in subjects with normal levels of CYP 2D6. Even so these levels were almost 5-fold lower than those seen in CYP 2D6 poor metabolizers.
- Coadministration of valdecoxib with phenytoin (a CYP 2C9/2C19 substrate) did not affect the pharmacokinetics of phenytoin.
- Coadministration of valdecoxib, or its injectable prodrug, with substrates of CYP 2C9 (propofol) and CYP 3A4 (midazolam, alfentanil, fentanyl) did not inhibit the metabolism of these substrates.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
- Valdecoxib was not carcinogenic in rats given oral doses up to 7.5 mg/kg/day for males and 1.5 mg/kg/day for females (equivalent to approximately 2- to 6-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)) or in mice given oral doses up to 25 mg/kg/day for males and 50 mg/kg/day for females (equivalent to approximately 0.6- to 2.4-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)) for two years.
- Valdecoxib was not mutagenic in an Ames test or a mutation assay in Chinese hamster ovary (CHO) cells, nor was it clastogenic in a chromosome aberration assay in CHO cells or in an in vivo micronucleus test in rat bone marrow.
- Valdecoxib did not impair male rat fertility at oral doses up to 9.0 mg/kg/day (equivalent to approximately 3- to 6-fold human exposure at 20 mg QD as measured by the AUC(0–24hr)). In female rats, a decrease in ovulation with increased pre- and post-implantation loss resulted in decreased live embryos/fetuses at doses ≥2 mg/kg/day (equivalent to approximately 2-fold human exposure at 20 mg QD as measured by the AUC(0–24hr) for valdecoxib). The effects on female fertility were reversible. This effect is expected with inhibition of prostaglandin synthesis and is not the result of irreversible alteration of female reproductive function.
Clinical Studies
- The efficacy and clinical utility of BEXTRA Tablets have been demonstrated in osteoarthritis (OA), rheumatoid arthritis (RA) and in the treatment of primary dysmenorrhea.
Osteoarthritis
- BEXTRA was evaluated for treatment of the signs and symptoms of osteoarthritis of the knee or hip, in five double-blind, randomized, controlled trials in which 3918 patients were treated for 3 to 6 months. BEXTRA was shown to be superior to placebo in improvement in three domains of OA symptoms: (1) the WOMAC (Western Ontario and McMaster Universities) osteoarthritis index, a composite of pain, stiffness and functional measures in OA, (2) the overall patient assessment of pain, and (3) the overall patient global assessment. The two 3-month pivotal trials in OA generally showed changes statistically significantly different from placebo, and comparable to the naproxen control, in measures of these domains for the 10 mg/day dose. No additional benefit was seen with a valdecoxib 20-mg daily dose.
Rheumatoid Arthritis
- BEXTRA demonstrated significant reduction compared to placebo in the signs and symptoms of RA, as measured by the ACR (American College of Rheumatology) 20 improvement, a composite defined as both improvement of 20% in the number of tender and number of swollen joints, and a 20% improvement in three of the following five: patient global, physician global, patient pain, patient function assessment, and C-reactive protein (CRP). BEXTRA was evaluated for treatment of the signs and symptoms of rheumatoid arthritis in four double-blind, randomized, controlled studies in which 3444 patients were treated for 3 to 6 months. The two 3-month pivotal trials compared valdecoxib to naproxen and placebo. The results for the ACR20 responses in these trials are shown below (Table 2). Trials of BEXTRA in rheumatoid arthritis allowed concomitant use of corticosteroids and/or disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate, gold salts, and hydroxychloroquine. No additional benefit was seen with a valdecoxib 20-mg daily dose.
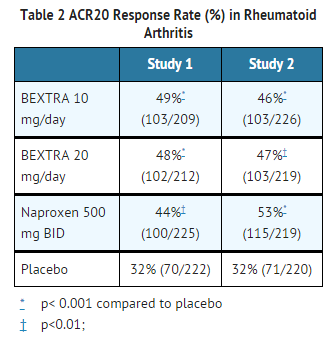
This image is provided by the National Library of Medicine.

Primary Dysmenorrhea
- BEXTRA was compared to naproxen sodium 550 mg in two placebo-controlled studies of women with moderate to severe primary dysmenorrhea. The onset of analgesia was within 60 minutes for BEXTRA 20 mg. The onset, magnitude, and duration of analgesic effect with BEXTRA 20 mg were comparable to naproxen sodium 550 mg.
Safety Studies
- Studies in post-surgical patients (Investigational use)
- Three placebo-controlled studies (two coronary artery bypass graft (CABG) surgery studies largely in patients with medial sternotomy placed on cardiopulmonary bypass and a single general surgery study) were conducted to evaluate the safety of the investigational agent, parecoxib sodium (the parenteral pro-drug of valdecoxib) and valdecoxib. Patients received parecoxib sodium for at least 3 days and then were transitioned to valdecoxib for a total treatment duration of 10–14 days. All patients received standard of care analgesia during treatment and all patients received low-dose aspirin prior to randomization and throughout the two CABG surgery studies.
- In addition to routine adverse event reporting, pre-specified adverse events of interest were adjudicated according to pre-specified definitions by an independent committee who were blinded to treatment assignment. In the three studies, the overall routine adverse event profiles were similar between active treatments and placebo.
- The first CABG surgery study evaluated patients treated with IV parecoxib sodium 40 mg bid for a minimum of 3 days, followed by treatment with valdecoxib 40 mg bid (parecoxib sodium/valdecoxib group) (n=311) or placebo/placebo (n=151) in a 14-day, double-blind placebo-controlled study. Nine pre-specified adverse event categories were evaluated (cardiovascular thromboembolic events, pericarditis, new onset or exacerbation of congestive heart failure, renal failure/dysfunction, upper GI ulcer complications, major non-GI bleeds, infections, non-infectious pulmonary complications, and death). There was a significantly (p<0.05) greater incidence of cardiovascular/thromboembolic events (myocardial infarction, ischemia, cerebrovascular accident, deep vein thrombosis and pulmonary embolism) detected in the parecoxib/valdecoxib treatment group compared to the placebo/placebo treatment group for the IV dosing period (2.2% and 0.0% respectively) and over the entire study period (4.8% and 1.3% respectively). Surgical wound complications (most involving the sternal wound) were observed at an increased rate with parecoxib/valdecoxib treatment.
- In the second larger CABG surgery study, four pre-specified event categories were evaluated (cardiovascular/thromboembolic; renal dysfunction/renal failure; upper GI ulcer/bleeding; surgical wound complication). Patients were randomized within 24-hours post-CABG surgery to: parecoxib initial dose of 40 mg IV, then 20 mg IV Q12H for a minimum of 3 days followed by valdecoxib PO (20 mg Q12H) (n=544) for the remainder of a 10 day treatment period; placebo IV followed by valdecoxib PO (n=544); or placebo IV followed by placebo PO (n=548). A significantly (p=0.033) greater incidence of events in the cardiovascular/thromboembolic category was detected in the parecoxib /valdecoxib treatment group (2.0%) compared to the placebo/placebo treatment group (0.5%). Placebo/valdecoxib treatment was also associated with a higher incidence of CV thromboembolic events versus placebo treatment, but this difference did not reach statistical significance. Three of the cardiovascular thromboembolic events in the placebo/valdecoxib treatment group occurred during the placebo treatment period; these patients did not receive valdecoxib. Pre-specified events that occurred with the highest incidence in all three treatment groups involved the category of surgical wound complications, including deep surgical infections and sternal wound healing events (see table below).
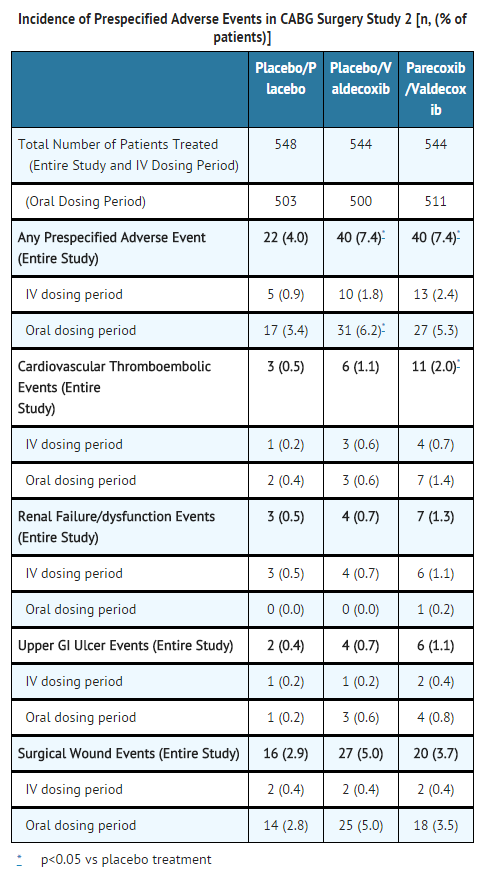
This image is provided by the National Library of Medicine.

- General Surgery: In the third study, a large (N=1050) major orthopedic/general surgery trial, patients received an initial dose of parecoxib 40 mg IV, then 20 mg IV Q12H for a minimum of 3 days followed by valdecoxib PO (20 mg Q12H) (n=525) for the remainder of a 10 day treatment period, or placebo IV followed by placebo PO (n=525). There were no significant differences in the overall safety profile, including the four pre-specified event categories described above for the second CABG surgery study, for parecoxib sodium/valdecoxib compared to placebo treatment in these post-surgical patients (see table below).
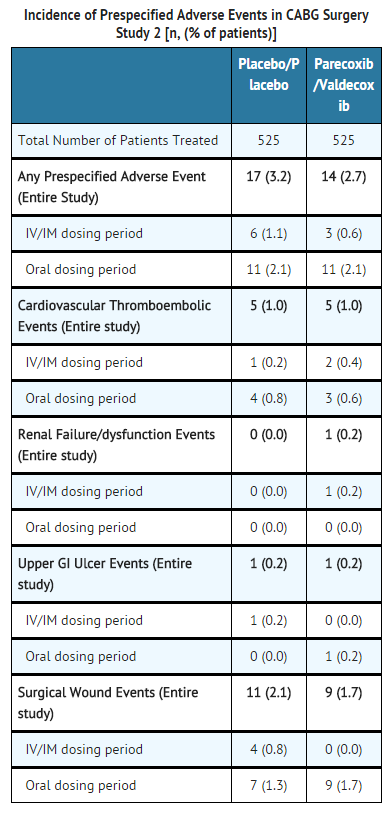
This image is provided by the National Library of Medicine.

- No significant differences were observed between the treatment groups
- BEXTRA is contraindicated for the treatment of post-operative pain immediately following coronary artery bypass graft surgery and should not be used in this setting .
- Cardiovascular Safety Analysis from Osteoarthritis and Rheumatoid Arthritis Studies
- Randomized controlled clinical trials with BEXTRA longer than one year have not been conducted, nor have studies powered to detect differences in cardiovascular events in a chronic setting been conducted.
- In an analysis of 10 randomized controlled clinical studies in osteoarthritis and rheumatoid arthritis, 4531 patients received BEXTRA in doses ranging from 10 mg to 80 mg for periods of 6 to 52 weeks. The majority of these patients received BEXTRA for 12 weeks or less. This analysis compared the incidence of serious cardiovascular events in BEXTRA-treated patients with the incidence of these events in patients receiving placebo (N=1142) or NSAID therapy (N=2261). In this analysis, no apparent differences were detected in the exposure-adjusted serious cardiovascular thromboembolic event rates between patients receiving BEXTRA, placebo and NSAIDs.
- BEXTRA has not been studied in clinical trials beyond 12 months duration.
- Gastrointestinal (GI) Endoscopy Studies with Therapeutic Doses
- Scheduled upper GI endoscopic evaluations were performed with BEXTRA at doses of 10 and 20 mg daily in over 800 OA patients who were enrolled into two randomized 3-month studies using active comparators and placebo controls (Study 3 and Study 4). These studies enrolled patients free of endoscopic ulcers at baseline and compared rates of endoscopic ulcers, defined as any gastroduodenal ulcer seen endoscopically provided it was of "unequivocal depth" and at least 3 mm in diameter.
- In both studies, BEXTRA 10 mg daily was associated with a statistically significant lower incidence of endoscopic gastroduodenal ulcers over the study period compared to the active comparators. Figure 1 summarizes the incidence of gastroduodenal ulcers in Studies 3 and 4 for the placebo, valdecoxib, and active control arms.
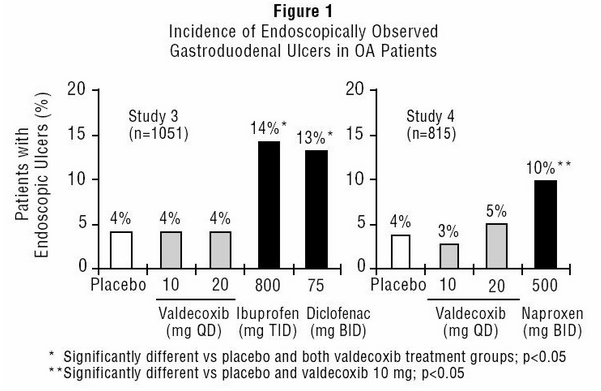
This image is provided by the National Library of Medicine.

- Safety Study with Supratherapeutic Doses
- Scheduled upper GI endoscopic evaluations were performed in a randomized 6-month study of 1217 patients with OA and RA comparing valdecoxib 20 mg BID (40 mg daily) and 40 mg BID (80 mg daily) (4 to 8 times the recommended therapeutic dose) to naproxen 500 mg BID (Study 5). This study also formally assessed renal events as a primary outcome with supratherapeutic doses of BEXTRA. The renal endpoint was defined as any of the following: new/increase in edema, new/increase in congestive heart failure, increase in blood pressure (BP; >20 mm Hg systolic, >10 mm Hg diastolic), new/increase in BP treatment, new/increase in diuretic therapy, creatinine increase over 30% (or >1.2 mg/dL if baseline <0.9 mg/dL), BUN increase over 200% or >50 mg/dL, 24-hr urinary protein increase to >500 mg (if baseline 0–150 mg or >750 if baseline 151–300 or >1000 if baseline 301–500), serum potassium increase to >6 mEq/L, or serum sodium decrease to <130 mEq/L.
- Figure 2 summarizes the incidence rates of gastroduodenal ulcers and renal events that were seen in Study 5. BEXTRA 40 mg daily and 80 mg daily were associated with a statistically significant lower incidence of endoscopic gastroduodenal ulcers over the study period compared to naproxen. The incidence of renal events was significantly different between the BEXTRA 80 mg daily group and naproxen. The clinical relevance of renal events observed with supratherapeutic doses (4 to 8 times the recommended therapeutic dose) of BEXTRA is not known
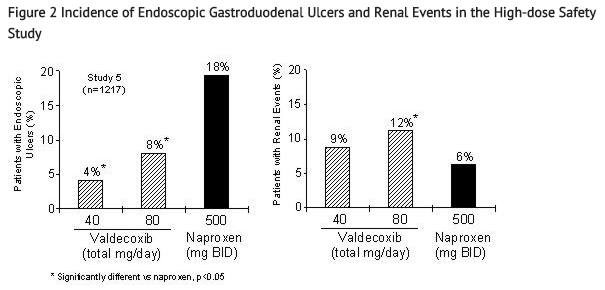
This image is provided by the National Library of Medicine.

- Renal Safety at the Therapeutic Chronic Dose
- The renal effects of valdecoxib compared with placebo and conventional NSAIDs were also assessed by prospectively designed pooled analyses of renal events data (see definition above —SUPRATHERAPEUTIC DOSES) from five placebo- and active-controlled 12-week arthritis trials that included 995 OA or RA patients given valdecoxib 10 mg daily. The incidence of renal events observed in this analysis with valdecoxib 10 mg daily (3%), ibuprofen 800 mg TID (7%), naproxen 500 mg BID (2%) and diclofenac 75 mg BID (4%) were significantly higher than placebo-treated patients (1%). In all treatment groups, the majority of renal events were either due to the occurrence of edema or worsening BP.
- Gastrointestinal Ulcers in High-Risk Patients
- Subset analyses were performed of patients with risk factors (age, concomitant low-dose aspirin use, history of prior ulcer disease) enrolled in four upper GI endoscopic studies. Table 3 summarizes the trends seen.
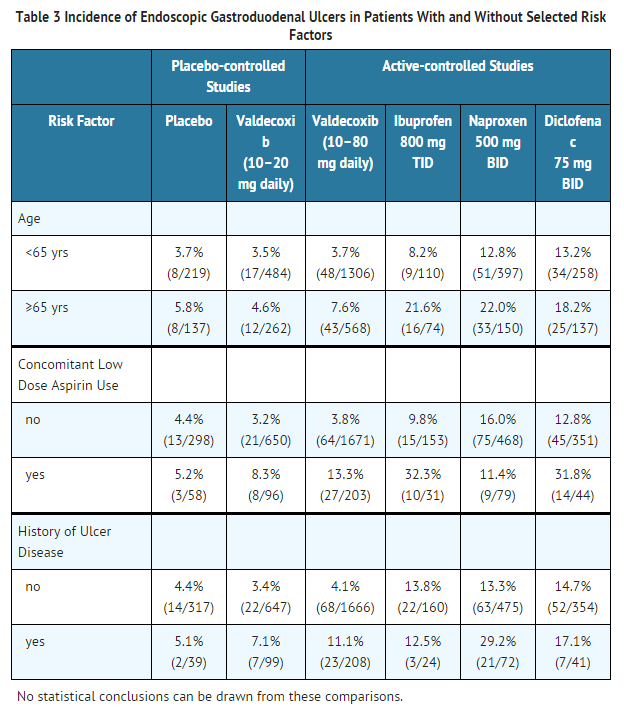
This image is provided by the National Library of Medicine.

- The correlation between findings of endoscopic studies, and the incidence of clinically significant serious upper GI events has not been established.
- In four clinical studies with young and elderly (≥65 years) subjects, single and multiple doses up to 7 days of BEXTRA 10 to 40 mg BID had no effect on platelet aggregation.
How Supplied
- BEXTRA Tablets 10 mg are white, film-coated, and capsule-shaped, debossed "10" on one side with a four pointed star shape on the other, supplied as:

- BEXTRA Tablets 20 mg are white, film-coated, and capsule-shaped, debossed "20" on one side with a four pointed star shape on the other, supplied as:

Storage
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F)
Images
Drug Images
{{#ask: Page Name::Valdecoxib |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
Ingredients and Appearance
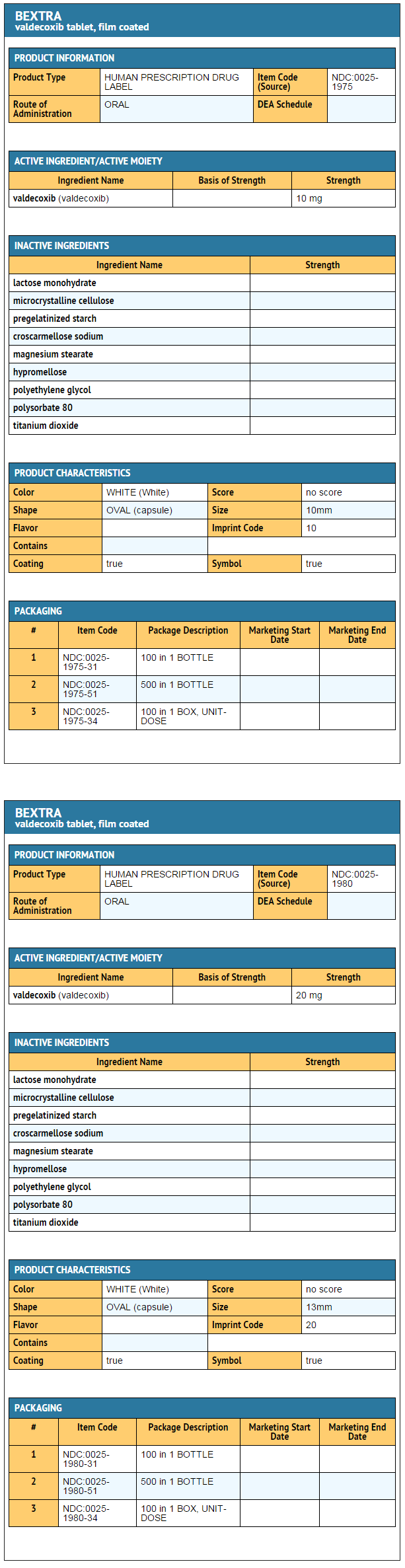
This image is provided by the National Library of Medicine.

{{#ask: Label Page::Valdecoxib |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- BEXTRA can cause GI discomfort and, rarely, more serious GI side effects, which may result in hospitalization and even fatal outcomes. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative sign or symptoms. Patients should be apprised of the importance of this follow-up (see WARNINGS — GASTROINTESTINAL (GI) EFFECTS — RISK OF GI ULCERATION, BLEEDING, AND PERFORATION).
- Patients should report to their physicians, signs or symptoms of gastrointestinal ulceration or bleeding, weight gain, or edema.
- Patients should be instructed to discontinue treatment and seek medical attention at the first signs of a skin reaction (pruritus, rash, erythema, or mucosal lesions) .
- Patients should also be instructed to seek immediate emergency help in the case of an anaphylactoid reaction.
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and flu-like symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical attention.
- In late pregnancy, BEXTRA should be avoided because it may cause premature closure of the ductus arteriosus.
Precautions with Alcohol
- Alcohol-Valdecoxib interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- Bextra®[1]
Look-Alike Drug Names
There is limited information regarding Valdecoxib Look-Alike Drug Names in the drug label.
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.