Tipranavir
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vignesh Ponnusamy, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
WARNING
See full prescribing information for complete Boxed Warning.
HEPATOTOXICITY and INTRACRANIAL HEMORRHAGE:
|
Overview
Tipranavir is a protease inhibitor, co-administered with ritonavir that is FDA approved for the treatment of HIV-1 infected patients who are treatment-experienced and infected with HIV-1 strains resistant to more than one protease inhibitor. There is a Black Box Warning for this drug as shown here. Common adverse reactions include diarrhea, nausea, pyrexia, vomiting, fatigue, headache, and abdominal pain.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
HIV-1 Infection
- APTIVUS must be co-administered with ritonavir to exert its therapeutic effect. Failure to correctly co-administer APTIVUS with ritonavir will result in plasma levels of tipranavir that will be insufficient to achieve the desired antiviral effect and will alter some drug interactions.
- The recommended adult dose of APTIVUS is 500 mg (two 250 mg capsules or 5 mL oral solution) co-administered with 200 mg of ritonavir, twice daily.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Tipranavir in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Tipranavir in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
HIV-1 Infection
- Healthcare professionals should pay special attention to accurate calculation of the dose of APTIVUS, transcription of the medication order, dispensing information and dosing instruction to minimize risk for medication errors, overdose, and underdose.
- Prescribers should calculate the appropriate dose of APTIVUS for each individual child based on body weight (kg) or body surface area (BSA, m2) and should not exceed the recommended adult dose.
- Before prescribing APTIVUS 250 mg capsules, children should be assessed for the ability to swallow capsules. If a child is unable to reliably swallow an APTIVUS capsule, the APTIVUS oral solution formulation should be prescribed.
- The recommended pediatric dose of APTIVUS is 14 mg/kg with 6 mg/kg ritonavir (or 375 mg/m2 co-administered with ritonavir 150 mg/m2) taken twice daily not to exceed a maximum dose of APTIVUS 500 mg co-administered with ritonavir 200 mg twice daily. For children who develop intolerance or toxicity and cannot continue with APTIVUS 14 mg/kg with 6 mg/kg ritonavir, physicians may consider decreasing the dose to APTIVUS 12 mg/kg with 5 mg/kg ritonavir (or APTIVUS 290 mg/m2 co-administered with 115 mg/m2 ritonavir) taken twice daily provided their virus is not resistant to multiple protease inhibitors.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Tipranavir in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Tipranavir in pediatric patients.
Contraindications
- Hepatic Impairment
- APTIVUS is contraindicated in patients with moderate or severe (Child-Pugh Class B or C, respectively) hepatic impairment.
- Drug Interactions
- Co-administration of APTIVUS/ritonavir with drugs that are highly dependent on CYP 3A for clearance or are potent CYP 3A inducers are contraindicated (see Table 1). These recommendations are based on either drug interaction studies or they are predicted interactions due to the expected magnitude of interaction and potential for serious events or loss of efficacy. For information regarding clinical recommendations.
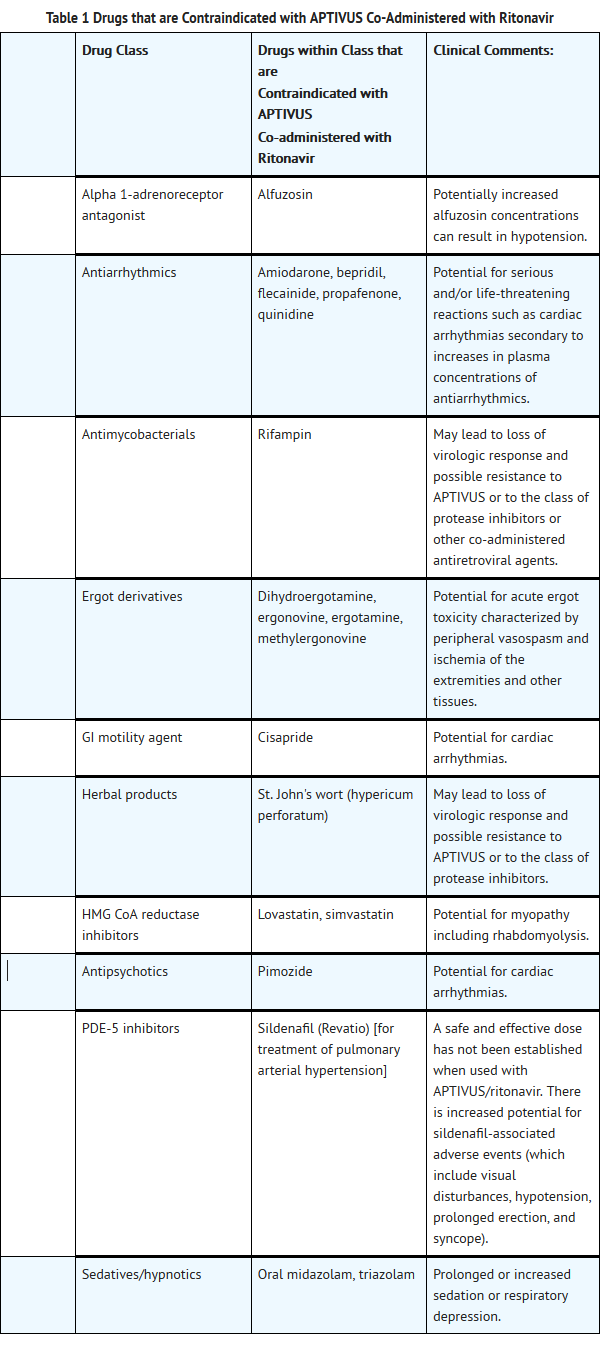
This image is provided by the National Library of Medicine.

Warnings
|
WARNING
See full prescribing information for complete Boxed Warning.
HEPATOTOXICITY and INTRACRANIAL HEMORRHAGE:
|
Precautions
- Hepatic Impairment and Toxicity
- Clinical hepatitis and hepatic decompensation, including some fatalities, were reported with APTIVUS co-administered with 200 mg of ritonavir. These have generally occurred in patients with advanced HIV-1 disease taking multiple concomitant medications. A causal relationship to APTIVUS/ritonavir could not be established. Physicians and patients should be vigilant for the appearance of signs or symptoms of hepatitis, such as fatigue, malaise, anorexia, nausea, jaundice, bilirubinuria, acholic stools, liver tenderness or hepatomegaly. Patients with signs or symptoms of clinical hepatitis should discontinue APTIVUS/ritonavir treatment and seek medical evaluation.
- All patients should be followed closely with clinical and laboratory monitoring, especially those with chronic hepatitis B or C co-infection, as these patients have an increased risk of hepatotoxicity. Liver function tests should be performed prior to initiating therapy with APTIVUS/ritonavir, and frequently throughout the duration of treatment.
- If asymptomatic elevations in AST or ALT greater than 10 times the upper limit of normal occur, APTIVUS/ritonavir therapy should be discontinued. If asymptomatic elevations in AST or ALT between 5 – 10 times the upper limit of normal and increases in total bilirubin greater than 2.5 times the upper limit of normal occur, APTIVUS/ritonavir therapy should be discontinued.
- Treatment-experienced patients with chronic hepatitis B or hepatitis C co-infection or elevated transaminases are at approximately 2-fold risk for developing Grade 3 or 4 transaminase elevations or hepatic decompensation. In two large, randomized, open-label, controlled clinical trials with an active comparator (1182.12 and 1182.48) of treatment-experienced patients, Grade 3 and 4 increases in hepatic transaminases were observed in 10.3% (10.9/100 PEY) receiving APTIVUS/ritonavir through week 48. In a study of treatment-naïve patients, 20.3% (21/100 PEY) experienced Grade 3 or 4 hepatic transaminase elevations while receiving APTIVUS/ritonavir 500 mg/200 mg through week 48.
- Tipranavir is principally metabolized by the liver. Caution should be exercised when administering APTIVUS/ritonavir to patients with mild hepatic impairment (Child-Pugh Class A) because tipranavir concentrations may be increased.
- Intracranial Hemorrhage
- APTIVUS, co-administered with 200 mg of ritonavir, has been associated with reports of both fatal and non-fatal intracranial hemorrhage (ICH). Many of these patients had other medical conditions or were receiving concomitant medications that may have caused or contributed to these events. No pattern of abnormal coagulation parameters has been observed in patients in general, or preceding the development of ICH. Therefore, routine measurement of coagulation parameters is not currently indicated in the management of patients on APTIVUS.
- Drug Interactions
- See Table 1 for a listing of contraindicated drugs with APTIVUS/ritonavir due to potentially life-threatening adverse events, significant drug interactions, or due to loss of virologic activity. See Table 4 for a listing of established and other potentially significant drug interactions with APTIVUS/ritonavir.
- Effects on Platelet Aggregation and Coagulation
- APTIVUS/ritonavir should be used with caution in patients who may be at risk of increased bleeding from trauma, surgery or other medical conditions, or who are receiving medications known to increase the risk of bleeding such as antiplatelet agents and anticoagulants, or who are taking supplemental high doses of vitamin E.
- In rats, tipranavir treatment alone induced dose-dependent changes in coagulation parameters, bleeding events and death. Co-administration with vitamin E significantly increased these effects. However, analyses of stored plasma from adult patients treated with APTIVUS capsules and pediatric patients treated with APTIVUS oral solution (which contains a vitamin E derivative) showed no effect of APTIVUS/ritonavir on vitamin K-dependent coagulation factors (Factor II and Factor VII), Factor V, or on prothrombin or activated partial thromboplastin times.
- In in vitro experiments, tipranavir was observed to inhibit human platelet aggregation at levels consistent with exposures observed in patients receiving APTIVUS/ritonavir.
- Vitamin E Intake
- Patients taking APTIVUS oral solution should be advised not to take supplemental vitamin E greater than a standard multivitamin as APTIVUS oral solution contains 116 IU/mL of vitamin E which is higher than the Reference Daily Intake (adults 30 IU, pediatrics approximately 10 IU).
- Rash
- Rash, including urticarial rash, maculopapular rash, and possible photosensitivity, has been reported in subjects receiving APTIVUS/ritonavir. In some cases rash was accompanied by joint pain or stiffness, throat tightness, or generalized pruritus. In controlled adult clinical trials, rash (all grades, all causality) was observed in 10% of females and in 8% of males receiving APTIVUS/ritonavir through 48 weeks of treatment. The median time to onset of rash was 53 days and the median duration of rash was 22 days. The discontinuation rate for rash in clinical trials was 0.5%. In an uncontrolled compassionate use program (n=3920), cases of rash, some of which were severe, accompanied by myalgia, fever, erythema, desquamation, and mucosal erosions were reported. In the pediatric clinical trial, the frequency of rash (all grades, all causality) through 48 weeks of treatment was 21%. Overall, most of the pediatric patients had mild rash and 5 (5%) had moderate rash. Overall 3% of pediatric patients interrupted APTIVUS treatment due to rash and the discontinuation rate for rash in pediatric patients was 0.9%. Discontinue and initiate appropriate treatment if severe skin rash develops.
- Sulfa Allergy
- APTIVUS should be used with caution in patients with a known sulfonamide allergy. Tipranavir contains a sulfonamide moiety. The potential for cross-sensitivity between drugs in the sulfonamide class and APTIVUS is unknown.
- Diabetes Mellitus/Hyperglycemia
- New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus and hyperglycemia have been reported during post-marketing surveillance in HIV-1 infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
- Immune Reconstitution Syndrome
- Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including APTIVUS. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia, tuberculosis, or reactivation of herpes simplex and herpes zoster), which may necessitate further evaluation and treatment.
- Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution, however, the time to onset is more variable, and can occur many months after initiation of treatment.
- Fat Redistribution
- Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
- Elevated Lipids
- Treatment with APTIVUS co-administered with 200 mg of ritonavir has resulted in large increases in the concentration of total cholesterol and triglycerides. Triglyceride and cholesterol testing should be performed prior to initiating APTIVUS/ritonavir therapy and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate; taking into account any potential drug-drug interactions.
- Patients with Hemophilia
- There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis in patients with hemophilia type A and B treated with protease inhibitors. In some patients additional Factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced if treatment had been discontinued. A causal relationship between protease inhibitors and these events has not been established.
- Resistance/Cross Resistance
- Because the potential for HIV-1 cross-resistance among protease inhibitors has not been fully explored in APTIVUS/ritonavir treated patients, it is unknown what effect therapy with APTIVUS will have on the activity of subsequently administered protease inhibitors.
Adverse Reactions
Clinical Trials Experience
- APTIVUS, co-administered with ritonavir, has been studied in a total of 6308 HIV-1 positive adults as combination therapy in clinical studies. Of these, 1299 treatment-experienced patients received the dose of 500/200 mg BID. Nine hundred nine (909) adults, including 541 in the 1182.12 and 1182.48 controlled clinical trials, have been treated for at least 48 weeks.
- In 1182.12 and 1182.48 in the APTIVUS/ritonavir arm, the most frequent adverse reactions were diarrhea, nausea, pyrexia, vomiting, fatigue, headache, and abdominal pain. The 48-Week Kaplan-Meier rates of adverse reactions leading to discontinuation were 13.3% for APTIVUS/ritonavir-treated patients and 10.8% for the comparator arm patients.
- Adverse reactions reported in the controlled clinical trials 1182.12 and 1182.48, based on treatment-emergent clinical adverse reactions of moderate to severe intensity (Grades 2 - 4) in at least 2% of treatment-experienced subjects in either treatment group are summarized in Table 2 below.
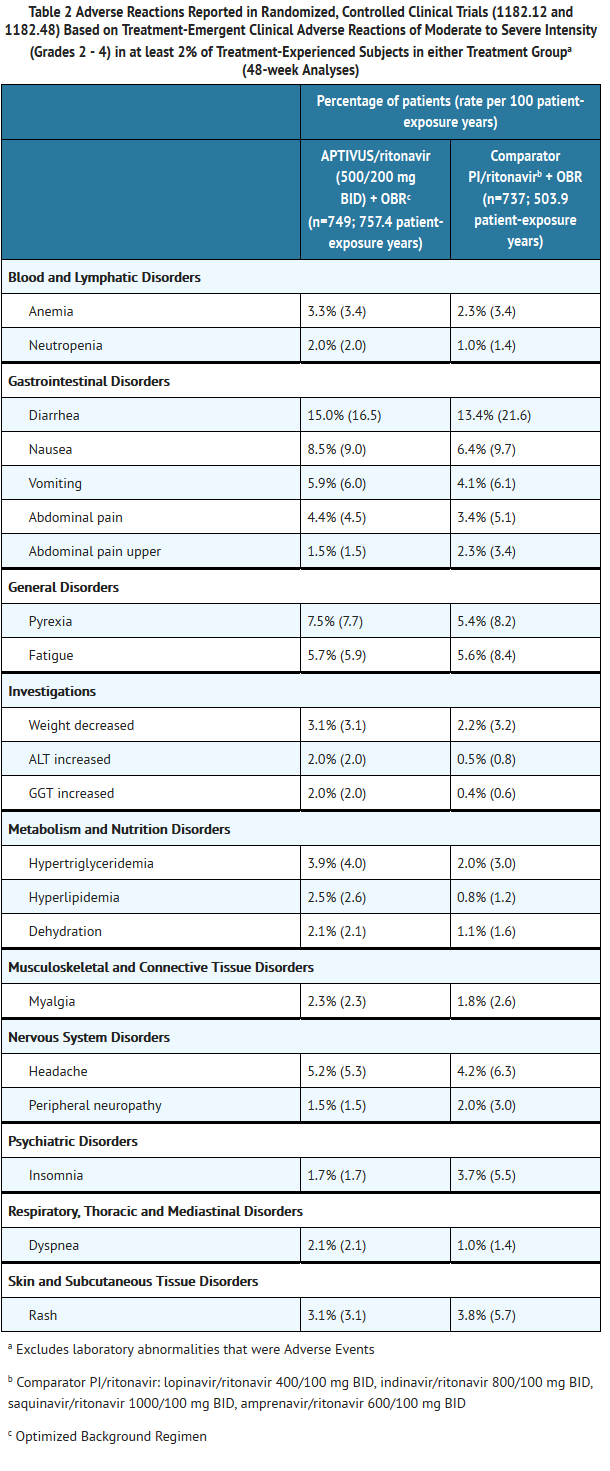
This image is provided by the National Library of Medicine.

- Less Common Adverse Reactions
- Other adverse reactions reported in <2% of adult patients (n=1474) treated with APTIVUS/ritonavir 500/200 mg in Phase 2 and 3 clinical trials are listed below by body system:
Blood and Lymphatic System Disorders
Gastrointestinal Disorders
Abdominal distension, dyspepsia, flatulence, gastroesophageal reflux disease, pancreatitis
General Disorders
Influenza-like illness, malaise
Hepatobiliary Disorders
Hepatitis, hepatic failure, hyperbilirubinemia, cytolytic hepatitis, toxic hepatitis, hepatic steatosis
Immune System Disorders
Investigations
Hepatic enzymes increased, liver function test abnormal, lipase increased
Metabolism and Nutrition Disorders
Anorexia, decreased appetite, diabetes mellitus, facial wasting, hyperamylasemia, hypercholesterolemia, hyperglycemia, mitochondrial toxicity
Musculoskeletal and Connective Tissue Disorders
Nervous System Disorders
Dizziness, intracranial hemorrhage, somnolence
Psychiatric Disorders
Renal and Urinary Disorders
Renal insufficiency
Skin and Subcutaneous System Disorders
Exanthem, lipoatrophy, lipodystrophy acquired, lipohypertrophy, pruritus
Laboratory Abnormalities
- Treatment-emergent laboratory abnormalities reported at 48 weeks in the controlled clinical trials 1182.12 and 1182.48 in adults are summarized in Table 3 below.
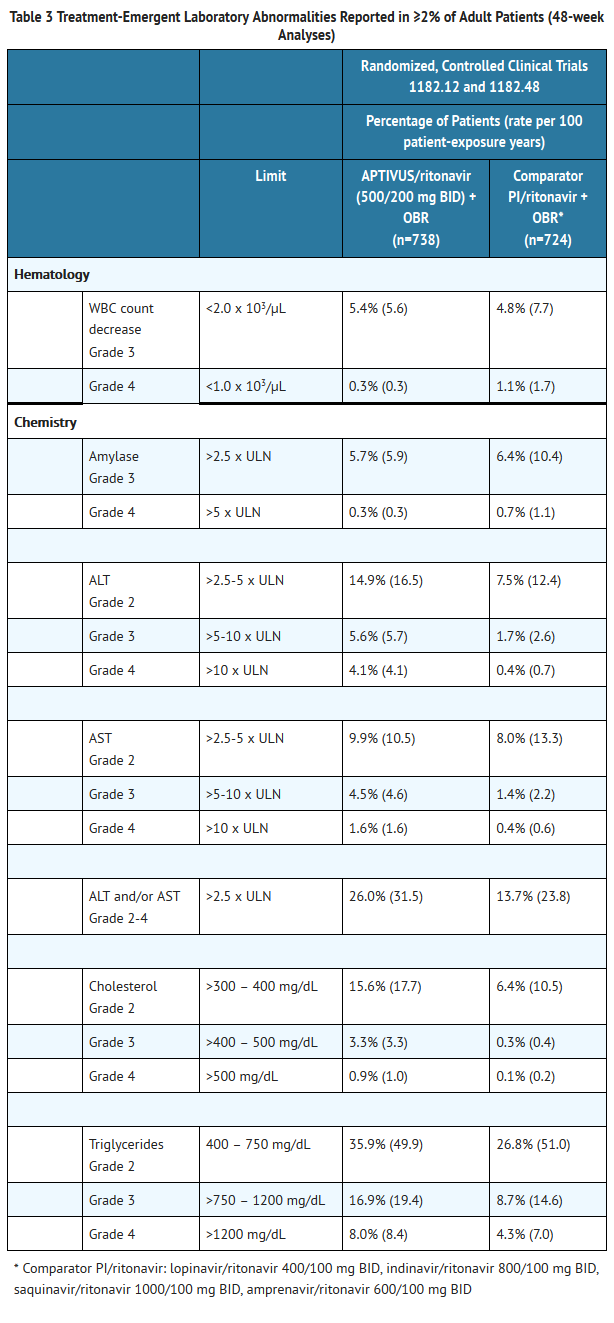
This image is provided by the National Library of Medicine.

Clinical Trials in Pediatric Patients
- APTIVUS, co-administered with ritonavir, has been studied in a total of 135 HIV-1 infected pediatric patients age 2 through 18 years as combination therapy. This study enrolled HIV-1 infected, treatment-experienced pediatric patients (with the exception of 3 treatment-naïve patients), with baseline HIV-1 RNA of at least 1500 copies/mL. One hundred and ten (110) patients were enrolled in a randomized, open-label 48-week clinical trial (Study 1182.14) and 25 patients were enrolled in other clinical studies including Expanded Access and Emergency Use Programs.
- The adverse reactions profile seen in Study 1182.14 was similar to adults. Pyrexia (6.4%), vomiting (5.5%), cough (5.5%), rash (5.5%), nausea (4.5%), and diarrhea (3.6%) were the most frequently reported adverse reactions (Grade 2-4, all causes) in pediatric patients. Rash was reported more frequently in pediatric patients than in adults.
- The most common Grade 3-4 laboratory abnormalities were increases in CPK (11%), ALT (6.5%), and amylase (7.5%).
- Due to previous reports of both fatal and non-fatal intracranial hemorrhage (ICH), an analysis of bleeding events was performed. At 48 weeks of treatment, the frequency of pediatric patients with any bleeding adverse reactions was 7.5%. No drug related serious bleeding adverse reaction was reported. The most frequent bleeding adverse reaction was epistaxis (3.7%). No other bleeding adverse reaction was reported in frequency of >1%. Additional trial follow-up through 100 weeks showed a cumulative 12% frequency of any bleeding adverse reaction.
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Tipranavir in the drug label.
Drug Interactions
- Potential for APTIVUS/ritonavir to Affect Other Drugs
- APTIVUS co-administered with ritonavir at the recommended dose is a net inhibitor of CYP 3A and may increase plasma concentrations of agents that are primarily metabolized by CYP 3A. Thus, co-administration of APTIVUS/ritonavir with drugs highly dependent on CYP 3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events is contraindicated. Co-administration with other CYP 3A substrates may require a dose adjustment or additional monitoring.
- Clinically significant drug-drug interactions of APTIVUS co-administered with ritonavir are summarized in Table 4 below.
- A phenotypic cocktail study was conducted with 16 healthy volunteers to quantify the influence of 10 days of APTIVUS/ritonavir capsule administration on the activity of hepatic CYP 1A2 (caffeine), 2C9 (warfarin), 2C19 (omeprazole), 2D6 (dextromethorphan) and the activity of intestinal and hepatic CYP 3A4/5 (midazolam) and P-glycoprotein (P-gp) (digoxin). This study determined the first-dose and steady-state effects of 500 mg of APTIVUS co-administered with 200 mg of ritonavir twice daily in capsule form. APTIVUS oral solution co-administered with ritonavir capsules demonstrated similar effects as APTIVUS capsules co-administrated with ritonavir.
- There was no net effect on CYP 2C9 or hepatic P-gp at first dose or steady state. There was no net effect after first dose on CYP 1A2, but there was moderate induction at steady state. There was modest inhibition of CYP 2C19 at the first dose, but there was marked induction at steady state. Potent inhibition of CYP 2D6 and both hepatic and intestinal CYP 3A4/5 activities were observed after first dose and steady state.
- Intestinal and hepatic P-gp activity was assessed by administering oral and intravenous digoxin, respectively. The digoxin results indicate P-gp was inhibited after the first dose of APTIVUS/ritonavir followed by induction of P-gp over time. Thus, it is difficult to predict the net effect of APTIVUS administered with ritonavir on oral bioavailability and plasma concentrations of drugs that are dual substrates of CYP 3A and P-gp. The net effect will vary depending on the relative affinity of the co-administered drugs for CYP 3A and P-gp, and the extent of intestinal first-pass metabolism/efflux. An in vitro induction study in human hepatocytes showed an increase in UGT1A1 by tipranavir similar to that evoked by rifampin. The clinical consequences of this finding have not been established.
- Potential for Other Drugs to Affect Tipranavir
- Tipranavir is a CYP 3A substrate and a P-gp substrate. Co-administration of APTIVUS/ritonavir and drugs that induce CYP 3A and/or P-gp may decrease tipranavir plasma concentrations. Co-administration of APTIVUS/ritonavir and drugs that inhibit P-gp may increase tipranavir plasma concentrations. Co-administration of APTIVUS/ritonavir with drugs that inhibit CYP 3A may not further increase tipranavir plasma concentrations, because the level of metabolites is low following steady-state administration of APTIVUS/ritonavir 500/200 mg twice daily.
- Clinically significant drug-drug interactions of APTIVUS co-administered with ritonavir are summarized in Table 4 below.

This image is provided by the National Library of Medicine.

Use in Specific Populations
Pregnancy
- Pregnancy Category C
- Investigation of fertility and early embryonic development with tipranavir disodium was performed in rats, teratogenicity studies were performed in rats and rabbits, and pre- and post-natal development were explored in rats.
- No teratogenicity was detected in reproductive studies performed in pregnant rats and rabbits up to dose levels of 1000 mg/kg/day and 150 mg/kg/day tipranavir, respectively, at exposure levels approximately 1.1-fold and 0.1-fold human exposure. At 400 mg/kg/day and above in rats, fetal toxicity (decreased sternebrae ossification and body weights) was observed, corresponding to an AUC of 1310 μM·h or approximately 0.8-fold human exposure at the recommended dose. In rats and rabbits, fetal toxicity was not noted at 40 mg/kg/day and 150 mg/kg/day, respectively, corresponding accordingly to Cmax/AUC0-24h levels of 30.4 μM/340 μM·h and 8.4 μM/120 μM·h. These exposure levels (AUC) are approximately 0.2-fold and 0.1-fold the exposure in humans at the recommended dose.
- In pre- and post-development studies in rats, tipranavir showed no adverse effects at 40 mg/kg/day (~0.2-fold human exposure), but caused growth inhibition in pups and maternal toxicity at dose levels of 400 mg/kg/day (~0.8-fold human exposure). No post-weaning functions were affected at any dose level.
- There are no adequate and well-controlled studies in pregnant women for the treatment of HIV-1 infection. APTIVUS should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Tipranavir in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Tipranavir during labor and delivery.
Nursing Mothers
- The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV-1. Because of both the potential for HIV-1 transmission and any possible adverse effects of APTIVUS, mothers should be instructed not to breast-feed if they are receiving APTIVUS.
Pediatric Use
- The safety, pharmacokinetic profile, and virologic and immunologic responses of APTIVUS oral solution and capsules were evaluated in HIV-1 infected pediatric patients age 2 to 18 years.
- The most frequent adverse reactions (grades 2-4) were similar to those described in adults. However, rash was reported more frequently in pediatric patients than in adults.
- The risk-benefit has not been established in pediatric patients <2 years of age.
Geriatic Use
- Clinical studies of APTIVUS/ritonavir did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently than younger subjects. In general, caution should be exercised in the administration and monitoring of APTIVUS in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Tipranavir with respect to specific gender populations.
Race
There is no FDA guidance on the use of Tipranavir with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Tipranavir in patients with renal impairment.
Hepatic Impairment
- Tipranavir is principally metabolized by the liver. Caution should be exercised when administering APTIVUS/ritonavir to patients with mild (Child-Pugh Class A) hepatic impairment because tipranavir concentrations may be increased. APTIVUS/ritonavir is contraindicated in patients with moderate or severe (Child-Pugh Class B or Child-Pugh Class C) hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Tipranavir in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Tipranavir in patients who are immunocompromised.
Administration and Monitoring
Administration
- Oral
Monitoring
There is limited information regarding Monitoring of Tipranavir in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Tipranavir in the drug label.
Overdosage
Acute Overdose
- There is no known antidote for APTIVUS overdose. Treatment of overdose should consist of general supportive measures, including monitoring of vital signs and observation of the patient’s clinical status. If indicated, elimination of unabsorbed tipranavir should be achieved by emesis or gastric lavage. Administration of activated charcoal may also be used to aid in removal of unabsorbed drug. Since tipranavir is highly protein bound, dialysis is unlikely to provide significant removal of the drug.
Chronic Overdose
There is limited information regarding Chronic Overdose of Tipranavir in the drug label.
Pharmacology


Mechanism of Action
- Tipranavir (TPV) is an HIV-1 protease inhibitor that inhibits the virus-specific processing of the viral Gag and Gag-Pol polyproteins in HIV-1 infected cells, thus preventing formation of mature virions.
- Antiviral Activity
- Tipranavir inhibits the replication of laboratory strains of HIV-1 and clinical isolates in acute models of T-cell infection, with 50% effective concentrations (EC50) ranging from 0.03 to 0.07 μM (18-42 ng/mL). Tipranavir demonstrates antiviral activity in cell culture against a broad panel of HIV-1 group M non-clade B isolates (A, C, D, F, G, H, CRF01 AE, CRF02 AG, CRF12 BF). Group O and HIV-2 isolates have reduced susceptibility in cell culture to tipranavir with EC50 values ranging from 0.164 -1 μM and 0.233-0.522 μM, respectively. When used with other antiretroviral agents in cell culture, the combination of tipranavir was additive to antagonistic with other protease inhibitors (amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir) and generally additive with the NNRTIs (delavirdine, efavirenz, and nevirapine) and the NRTIs (abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zidovudine). Tipranavir was synergistic with the HIV-1 fusion inhibitor enfuvirtide. There was no antagonism of the cell culture combinations of tipranavir with either adefovir or ribavirin, used in the treatment of viral hepatitis.
Structure
- APTIVUS is a protease inhibitor of HIV-1 belonging to the class of 4-hydroxy-5,6-dihydro-2-pyrone sulfonamides.
- The chemical name of tipranavir is 2-Pyridinesulfonamide, N-[3-[(1R)-1-[(6R)-5,6-dihydro-4-hydroxy-2-oxo-6-(2-phenylethyl)-6-propyl-2H-pyran-3-yl]propyl]phenyl]-5-(trifluoromethyl). It has a molecular formula of C31H33F3N2O5S and a molecular weight of 602.7. Tipranavir has the following structural formula and is a single stereoisomer with the 1R, 6R configuration.
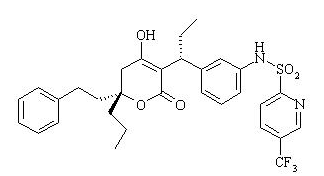
This image is provided by the National Library of Medicine.

- Tipranavir is a white to off-white to slightly yellow solid. It is freely soluble in dehydrated alcohol and propylene glycol, and insoluble in aqueous buffer at pH 7.5.
- APTIVUS soft gelatin capsules are for oral administration. Each capsule contains 250 mg tipranavir. The major inactive ingredients in the capsule are dehydrated alcohol (7% w/w or 0.1 g per capsule), polyoxyl 35 castor oil, propylene glycol, mono/diglycerides of caprylic/capric acid and gelatin.
- APTIVUS oral solution is available in a strength of 100 mg/mL of tipranavir. APTIVUS oral solution is a yellow, viscous clear liquid with a buttermint-butter toffee flavor. The major inactive ingredients in the oral solution are polyethylene glycol 400, vitamin E polyethylene glycol succinate (TPGS), purified water, and propylene glycol. Each milliliter of APTIVUS oral solution contains 116 IU of vitamin E, and when taken at the recommended maximum dose of 500 mg/200 mg tipranavir/ritonavir BID results in a daily dose of 1160 IU.
Pharmacodynamics
- ECG Evaluation
- The effect of APTIVUS/ritonavir on the QTcF interval was measured in a study in which 81 healthy subjects received the following treatments twice daily for 2.5 days: APTIVUS/ritonavir (500/200 mg), APTIVUS/ritonavir at a supra-therapeutic dose (750/200 mg), and placebo/ritonavir (-/200 mg). After baseline and placebo adjustment, the maximum mean QTcF change was 3.2 ms (1-sided 95% Upper CI: 5.6 ms) for the 500/200 mg dose and 8.3 ms (1-sided 95% Upper CI: 10.9 ms) for the supra-therapeutic 750/200 mg dose.
- Antiviral Activity in vivo
- The median Inhibitory Quotient (IQ) determined from 264 treatment-experienced adult patients was about 80 (inter-quartile range: 31-226), from the controlled clinical trials 1182.12 and 1182.48. The IQ is defined as the tipranavir trough concentration divided by the viral EC50 value, corrected for protein binding. There was a relationship between the proportion of patients with a ≥1 log10 reduction of viral load from baseline at week 48 and their IQ value. Among the 198 patients receiving APTIVUS/ritonavir with no new enfuvirtide use (e.g., new enfuvirtide, defined as initiation of enfuvirtide for the first time), the response rate was 23% in those with an IQ value <80 and 59% in those with an IQ value ≥80. Among the 66 patients receiving APTIVUS/ritonavir with new enfuvirtide, the response rates in patients with an IQ value <80 versus those with an IQ value ≥80 were 55% and 71%, respectively. These IQ groups are derived from a select population and are not meant to represent clinical breakpoints.
Pharmacokinetics
- In order to achieve effective tipranavir plasma concentrations and a twice-daily dosing regimen, co-administration of APTIVUS with ritonavir is essential [see Dosage and Administration (2)]. Ritonavir inhibits hepatic cytochrome P450 3A (CYP 3A), the intestinal P-gp efflux pump and possibly intestinal CYP 3A. In a dose-ranging evaluation in 113 HIV-1 negative male and female volunteers, there was a 29-fold increase in the geometric mean morning steady-state trough plasma concentrations of tipranavir following APTIVUS co-administered with low-dose ritonavir (500/200 mg twice daily) compared to APTIVUS 500 mg twice daily without ritonavir. In adults the mean systemic ritonavir concentration when 200 mg of ritonavir was given with 500 mg of APTIVUS was similar to the concentrations observed when 100 mg was given with the other protease inhibitors.
- Figure 1 displays mean plasma concentrations of tipranavir and ritonavir at steady state for 30 HIV-1 infected adult patients dosed with 500/200 mg tipranavir/ritonavir for 14 days.
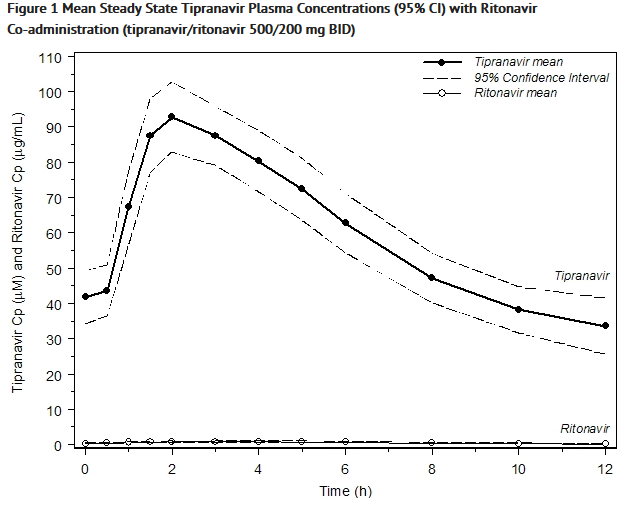
This image is provided by the National Library of Medicine.

- Absorption and Bioavailability
- Absorption of tipranavir in humans is limited, although no absolute quantification of absorption is available. Tipranavir is a P-gp substrate, a weak P-gp inhibitor, and appears to be a potent P-gp inducer as well. In vivo data suggest that tipranavir/ritonavir, at the dose of 500/200 mg, is a P-gp inhibitor after the first dose and induction of P-gp occurs over time. Tipranavir trough concentrations at steady-state are about 70% lower than those on Day 1, presumably due to intestinal P-gp induction. Steady state is attained in most subjects after 7-10 days of dosing.
- Dosing APTIVUS 500 mg with 200 mg ritonavir capsules twice daily for greater than 2 weeks and without meal restriction produced the pharmacokinetic parameters for male and female HIV-1 positive patients presented in Table 5.
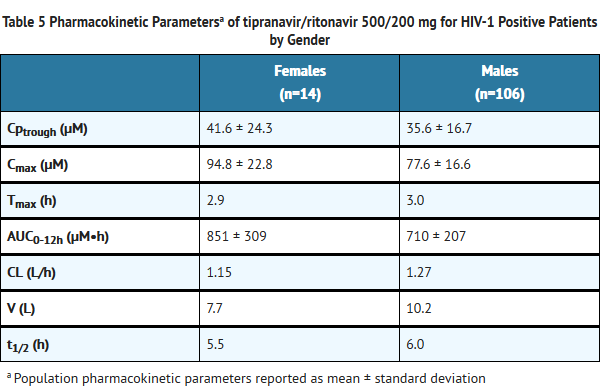
This image is provided by the National Library of Medicine.

- Effects of Food on Oral Absorption
- For APTIVUS capsules or oral solution co-administered with ritonavir capsules at steady-state, no clinically significant changes in tipranavir Cmax, Cp12h, and AUC were observed under fed conditions (500-682 Kcal, 23-25% calories from fat) compared to fasted conditions [see Dosage and Administration (2)]. The effect of food on tipranavir exposure when APTIVUS capsules or oral solution is co-administered with ritonavir tablets has not been evaluated [see Dosage and Administration (2)]. For information on the effect of food on the bioavailability of ritonavir tablets, please refer to the ritonavir tablet prescribing information.
- Distribution
- Tipranavir is extensively bound to plasma proteins (>99.9%). It binds to both human serum albumin and α-1-acid glycoprotein. The mean fraction of tipranavir (dosed without ritonavir) unbound in plasma was similar in clinical samples from healthy volunteers and HIV-1 positive patients. Total plasma tipranavir concentrations for these samples ranged from 9 to 82 μM. The unbound fraction of tipranavir appeared to be independent of total drug concentration over this concentration range.
- No studies have been conducted to determine the distribution of tipranavir into human cerebrospinal fluid or semen.
- Metabolism
- In vitro metabolism studies with human liver microsomes indicated that CYP 3A4 is the predominant CYP enzyme involved in tipranavir metabolism.
- The oral clearance of tipranavir decreased after the addition of ritonavir, which may represent diminished first-pass clearance of the drug at the gastrointestinal tract as well as the liver.
- The metabolism of tipranavir in the presence of 200 mg ritonavir is minimal. Administration of 14C-tipranavir to subjects that received APTIVUS/ritonavir 500/200 mg dosed to steady-state demonstrated that unchanged tipranavir accounted for 98.4% or greater of the total plasma radioactivity circulating at 3, 8, or 12 hours after dosing. Only a few metabolites were found in plasma, and all were at trace levels (0.2% or less of the plasma radioactivity). In feces, unchanged tipranavir represented the majority of fecal radioactivity (79.9% of fecal radioactivity). The most abundant fecal metabolite, at 4.9% of fecal radioactivity (3.2% of dose), was a hydroxyl metabolite of tipranavir. In urine, unchanged tipranavir was found in trace amounts (0.5% of urine radioactivity). The most abundant urinary metabolite, at 11.0% of urine radioactivity (0.5% of dose) was a glucuronide conjugate of tipranavir.
- Elimination
- Administration of 14C-tipranavir to subjects (n=8) that received APTIVUS/ritonavir 500/200 mg dosed to steady-state demonstrated that most radioactivity (median 82.3%) was excreted in feces, while only a median of 4.4% of the radioactive dose administered was recovered in urine. In addition, most radioactivity (56%) was excreted between 24 and 96 hours after dosing. The effective mean elimination half-life of tipranavir/ritonavir in healthy volunteers (n=67) and HIV-1 infected adult patients (n=120) was approximately 4.8 and 6.0 hours, respectively, at steady state following a dose of 500/200 mg twice daily with a light meal.
- Special Populations
- Renal Impairment
- APTIVUS pharmacokinetics have not been studied in patients with renal dysfunction. However, since the renal clearance of tipranavir is negligible, a decrease in total body clearance is not expected in patients with renal insufficiency.
- Hepatic Impairment
- In a study comparing 9 HIV-1 negative patients with mild (Child-Pugh Class A) hepatic impairment to 9 HIV-1 negative controls, the single and multiple dose plasma concentrations of tipranavir and ritonavir were increased in patients with hepatic impairment, but were within the range observed in clinical trials. No dosing adjustment is required in patients with mild hepatic impairment.
- The influence of moderate hepatic impairment (Child-Pugh Class B) or severe hepatic impairment (Child-Pugh Class C) on the multiple-dose pharmacokinetics of tipranavir administered with ritonavir has not been evaluated [see Dosage and Administration (2), Contraindications (4.1), and Warnings and Precautions (5.1)].
- Gender
- Evaluation of steady-state plasma tipranavir trough concentrations at 10-14 h after dosing from the controlled clinical trials 1182.12 and 1182.48 demonstrated that females generally had higher tipranavir concentrations than males. After 4 weeks of APTIVUS/ritonavir 500/200 mg BID, the median plasma trough concentration of tipranavir was 43.9 μM for females and 31.1 μM for males. The difference in concentrations does not warrant a dose adjustment.
- Race
- Evaluation of steady-state plasma tipranavir trough concentrations at 10-14 h after dosing from the controlled clinical trials 1182.12 and 1182.48 demonstrated that white males generally had more variability in tipranavir concentrations than black males, but the median concentration and the range making up the majority of the data are comparable between the races.
- Geriatric Patients
- Evaluation of steady-state plasma tipranavir trough concentrations at 10-14 h after dosing from the controlled clinical trials 1182.12 and 1182.48 demonstrated that there was no change in median trough tipranavir concentrations as age increased for either gender through 65 years of age. There were an insufficient number of women greater than age 65 years in the two trials to evaluate the elderly.
- Pediatric Patients
- Among pediatric patients in clinical trial 1182.14, steady-state plasma tipranavir trough concentrations were obtained 10 to 14 hours following study drug administration. Pharmacokinetic parameters by age group are presented in Table 6.
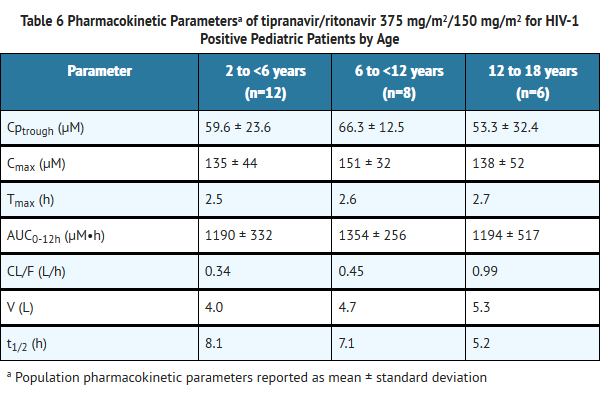
This image is provided by the National Library of Medicine.

- Drug Interactions
- Drug interaction studies were performed with APTIVUS capsules co-administered with ritonavir, and other drugs likely to be co-administered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of co-administration of APTIVUS with 200 mg ritonavir on the AUC, Cmax, and Cmin of tipranavir or the co-administered drug, are summarized in Tables 7 and 8, respectively. For information regarding clinical recommendations see Drug Interactions (7.2).
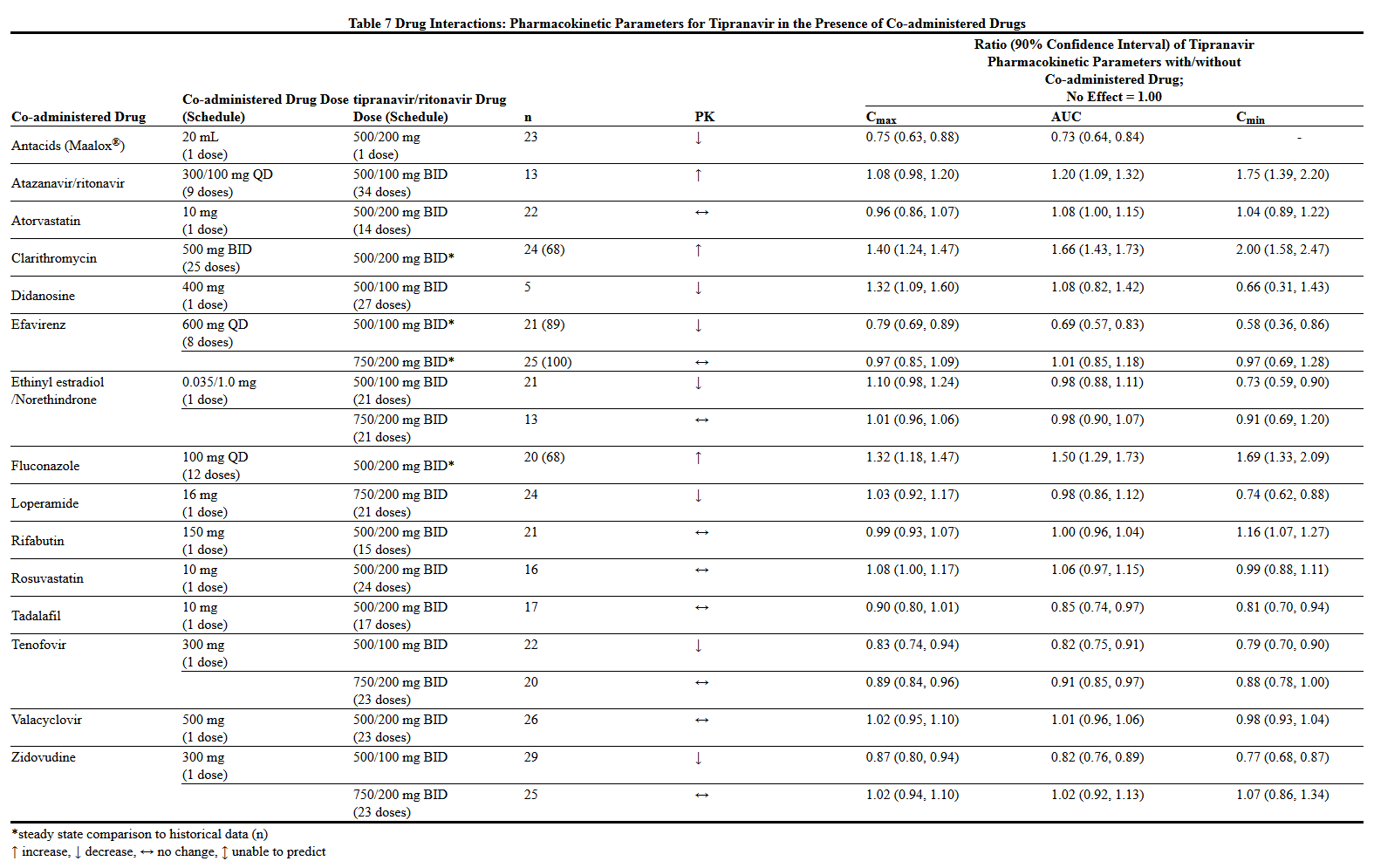
This image is provided by the National Library of Medicine.

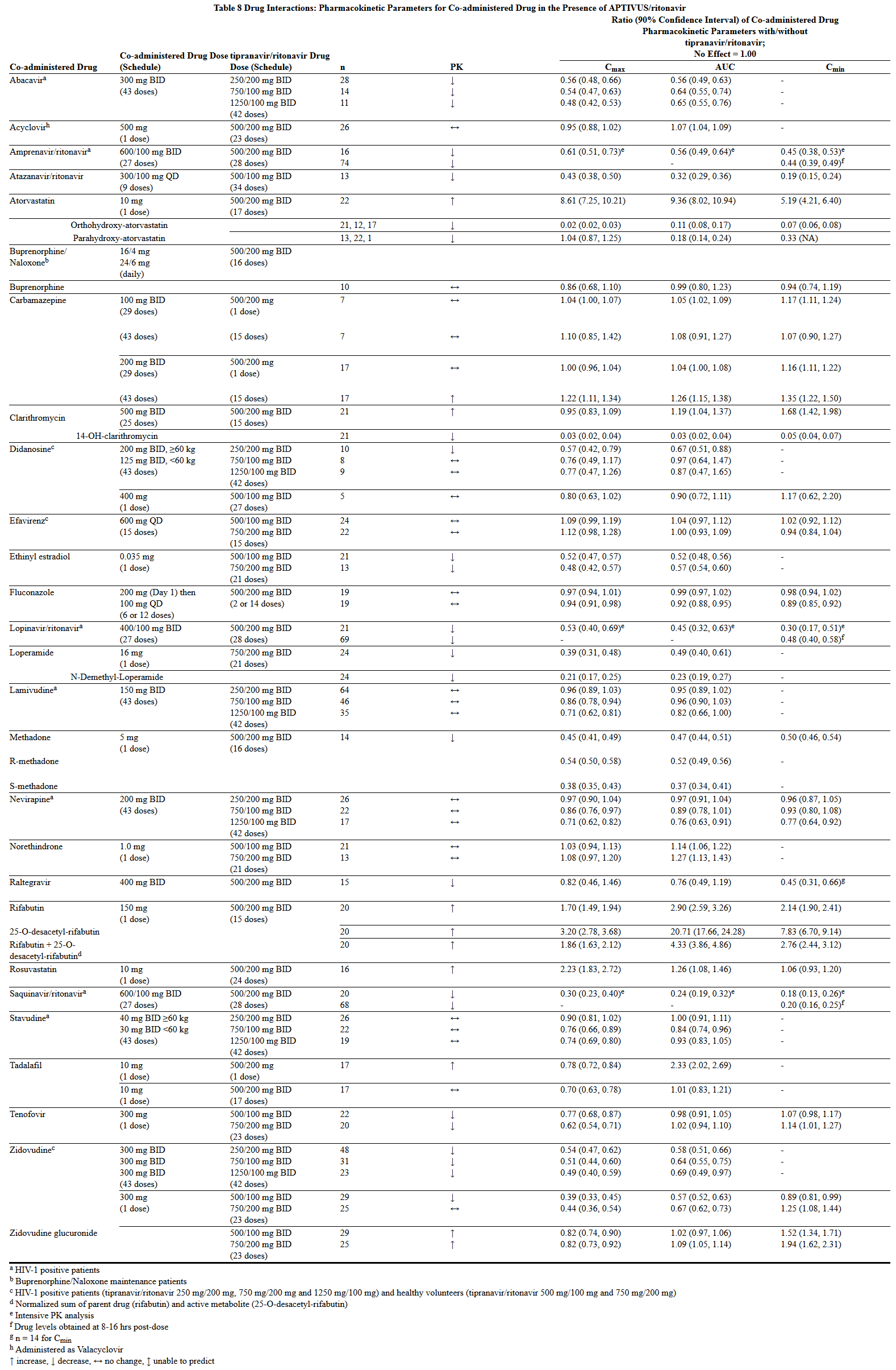
This image is provided by the National Library of Medicine.

Nonclinical Toxicology
- Long-term carcinogenicity studies in mice and rats have been conducted with tipranavir. Mice were administered 30, 150 or 300 mg/kg/day tipranavir, 150/40 mg/kg/day tipranavir/ritonavir in combination, or 40 mg/kg/day ritonavir. The incidences of benign hepatocellular adenomas and combined adenomas/carcinomas were increased in females of all groups except the low dose of tipranavir. These tumors were also increased in male mice at the high-dose of tipranavir and the tipranavir/ritonavir combination group. Hepatocellular carcinoma incidence was increased in female mice given the high dose of tipranavir and both sexes receiving tipranavir/ritonavir. The combination of tipranavir and ritonavir caused an exposure-related increase in this same tumor type in both sexes. The clinical relevance of the carcinogenic findings in mice is unknown. Systemic exposures in mice (based on AUC or Cmax) at all dose levels tested were below those in humans receiving the recommended dose level. Rats were administered 30, 100 or 300 mg/kg/day tipranavir, 100/26.7 mg/kg/day tipranavir/ritonavir in combination, or 10 mg/kg/day ritonavir. No drug-related findings in male rats were observed. At the highest dose of tipranavir, an increased incidence of benign follicular cell adenomas of the thyroid gland was observed in female rats. Based on AUC measurements, exposure to tipranavir at this dose level in rats is approximately equivalent to exposure in humans at the recommended therapeutic dose. This finding is probably not relevant to humans, because thyroid follicular cell adenomas are considered a rodent-specific effect secondary to enzyme induction.
- Tipranavir showed no evidence of mutagenicity or clastogenicity in a battery of five in vitro and in vivo tests including the Ames bacterial reverse mutation assay using S. typhimurium and E. coli, unscheduled DNA synthesis in rat hepatocytes, induction of gene mutation in Chinese hamster ovary cells, a chromosome aberration assay in human peripheral lymphocytes, and a micronucleus assay in mice.
- Tipranavir had no effect on fertility or early embryonic development in rats at dose levels up to 1000 mg/kg/day, equivalent to a Cmax of 258 μM in females. Based on Cmax levels in these rats, as well as an exposure (AUC) of 1670 μM·h in pregnant rats from another study, this exposure was approximately equivalent to the anticipated exposure in humans at the recommended dose level of 500/200 mg APTIVUS/ritonavir BID.
Clinical Studies
Adult Patients
- The following clinical data is derived from analyses of 48-week data from ongoing studies measuring effects on plasma HIV-1 RNA levels and CD4+ cell counts. At present there are no results from controlled studies evaluating the effect of APTIVUS/ritonavir on clinical progression of HIV-1.
- APTIVUS/ritonavir 500/200 mg BID + optimized background regimen (OBR) vs. Comparator Protease Inhibitor/ritonavir BID + OBR
- The two clinical trials 1182.12 and 1182.48 (RESIST 1 and RESIST 2) are ongoing, randomized, controlled, open-label, multicenter studies in HIV-1 positive, triple antiretroviral class experienced patients. All patients were required to have previously received at least two protease inhibitor-based antiretroviral regimens and were failing a protease inhibitor-based regimen at the time of study entry with baseline HIV-1 RNA at least 1000 copies/mL and any CD4+ cell count. At least one primary protease gene mutation from among 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V or 90M had to be present at baseline, with not more than two mutations at codons 33, 82, 84 or 90.
- These studies evaluated treatment response at 48 weeks in a total of 1483 patients receiving either APTIVUS co-administered with 200 mg of ritonavir plus OBR versus a control group receiving a ritonavir-boosted protease inhibitor (lopinavir, amprenavir, saquinavir or indinavir) plus OBR. Prior to randomization, OBR was individually defined for each patient based on genotypic resistance testing and patient history. The investigator had to declare OBR, comparator protease inhibitor, and use of new enfuvirtide prior to randomization. Randomization was stratified by choice of comparator protease inhibitor and use of new enfuvirtide.
- After Week 8, patients in the control group who met the protocol defined criteria of initial lack of virologic response or confirmed virologic failure had the option of discontinuing treatment and switching to APTIVUS/ritonavir in a separate roll-over study.
- Demographics and baseline characteristics were balanced between the APTIVUS/ritonavir arm and control arm. In both studies combined, the 1483 patients had a median age of 43 years (range 17-80), and were 86.3% male, 75.6% white, 12.9% black, and 0.9% Asian. The median baseline plasma HIV-1 RNA for both treatment groups was 4.8 (range 2.0 to 6.8) log10 copies/mL and median baseline CD4+ cell count was 162 (range 1 to 1894) cells/mm3. Overall, 38.4% of patients had a baseline HIV-1 RNA of >100,000 copies/mL, 58.6% had a baseline CD4+ cell count ≤200 cells/mm3, and 57.8% had experienced an AIDS defining Class C event at baseline.
- Patients had prior exposure to a median of 6 NRTIs, 1 NNRTI, and 4 PIs. A total of 10.1% of patients had previously used enfuvirtide. In baseline patient samples (n=454), 97% of the HIV-1 isolates were resistant to at least one protease inhibitor, 95% of the isolates were resistant to at least one NRTI, and >75% of the isolates were resistant to at least one NNRTI.
- The individually pre-selected protease inhibitor based on genotypic testing and the patient’s medical history was lopinavir in 48.7%, amprenavir in 26.4%, saquinavir in 21.8% and indinavir in 3.1% of patients. A total of 85.1% were possibly resistant or resistant to the pre-selected comparator protease inhibitors. Approximately 21% of patients used enfuvirtide during the study of which 16.6% in the APTIVUS/ritonavir arm and 13.2% in the comparator/ritonavir arm represented first time use of enfuvirtide (new enfuvirtide).
- Treatment response and efficacy outcomes of randomized treatment through Week 48 of studies 1182.12 and 1182.48 are shown in Table 12.
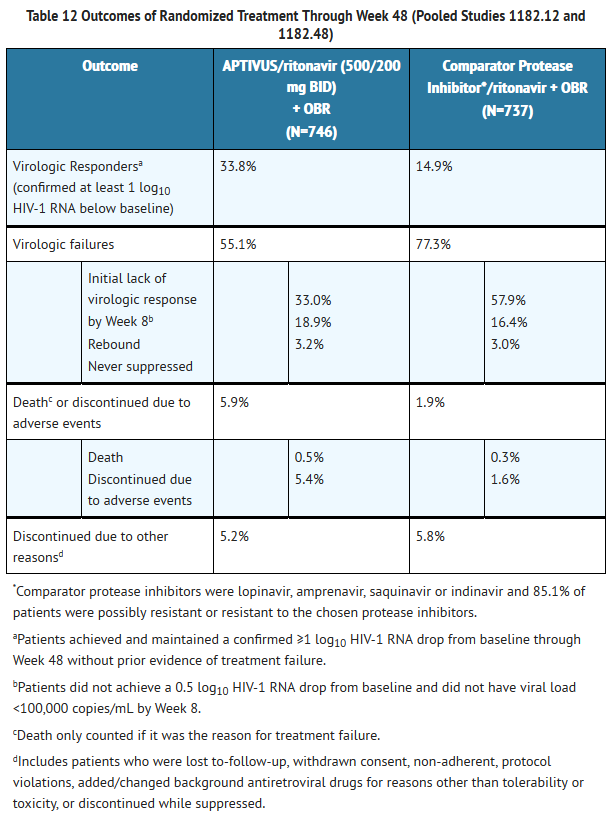
This image is provided by the National Library of Medicine.

- Through 48 weeks of treatment, the proportion of patients in the APTIVUS/ritonavir arm compared to the comparator PI/ritonavir arm with HIV-1 RNA <400 copies/mL was 30.3% and 13.6% respectively, and with HIV-1 RNA <50 copies/mL was 22.7% and 10.2% respectively. Among all randomized and treated patients, the median change from baseline in HIV-1 RNA at the last measurement up to Week 48 was -0.64 log10 copies/mL in patients receiving APTIVUS/ritonavir versus -0.22 log10 copies/mL in the comparator PI/ritonavir arm.
- Among all randomized and treated patients, the median change from baseline in CD4+ cell count at the last measurement up to Week 48 was +23 cells/mm3 in patients receiving APTIVUS/ritonavir (N=740) versus +4 cells/mm3 in the comparator PI/ritonavir (N=727) arm.
- Patients in the APTIVUS/ritonavir arm achieved a significantly better virologic outcome when APTIVUS/ritonavir was combined with enfuvirtide. Among patients with new enfuvirtide use, the proportion of patients in the APTIVUS/ritonavir arm compared to the comparator PI/ritonavir arm with HIV-1 RNA <400 copies/mL was 52.4% and 19.6% respectively, and with HIV-1 RNA <50 copies/mL was 37.3% and 14.4% respectively [see Clinical Pharmacology (12.2, 12.4)]. The median change from baseline in CD4+ cell count at the last measurement up to Week 48 was +89 cells/mm3 in patients receiving APTIVUS/ritonavir in combination with newly introduced enfuvirtide (N=124) and +18 cells/mm3 in the comparator PI/ritonavir (N=96) arm.
Pediatric Patients
- The pharmacokinetic profile, safety and activity of APTIVUS/ritonavir was evaluated in a randomized, open-label, multicenter study. This study enrolled HIV-1 infected, treatment-experienced pediatric patients (with the exception of 3 treatment-naïve patients), with baseline HIV-1 RNA of at least 1500 copies/mL. The age ranged from 2 through 18 years and patients were stratified by age (2 to <6 years, 6 to <12 years and 12 to 18 years). One hundred and ten (110) patients were randomized to receive one of two APTIVUS/ritonavir dose regimens: 375 mg/m2/150 mg/m2 dose (N=55) or 290 mg/m2/115 mg/m2 dose (N=55), plus background therapy of at least two non-protease inhibitor antiretroviral drugs, optimized using baseline genotypic resistance testing. All patients initially received APTIVUS oral solution. Pediatric patients who were 12 years or older and received the maximum dose of 500/200 mg BID could subsequently change to APTIVUS capsules at day 28 [see Adverse Reactions (6.2), Use in Specific Populations (8.4), Clinical Pharmacology (12.3), and Microbiology (12.4)].
- Demographics and baseline characteristics were balanced between the APTIVUS/ritonavir dose groups. The 110 randomized pediatric patients had a median age of 11.7 years (range 2 to 18), and were 57.2% male, 68.1% white, 30% black, and 1.8% Asian. The median baseline plasma HIV-1 RNA was 4.7 (range 3.0 to 6.8) log10 copies/mL and median baseline CD4+ cell count was 379 (range 2 to 2578) cells/mm3. Overall, 37.4% of patients had a baseline HIV-1 RNA of >100,000 copies/mL; 28.7% had a baseline CD4+ cell count ≤200 cells/mm3, and 48% had experienced a prior AIDS defining Class C event at baseline. Patients had prior exposure to a median of 4 NRTIs, 1 NNRTI, and 2 PIs.
- Eighty three (75%) completed the 48 week period while 25% discontinued prematurely. Of the patients who discontinued prematurely, 9 (8%) discontinued due to virologic failure, and 9 (8%) discontinued due to adverse reactions.
- At 48 weeks, 40% of patients had viral load <400 copies/mL. The proportion of patients with viral load <400 copies/mL tended to be greater (70%) in the youngest group of patients, who had less baseline viral resistance, compared to the older groups (37% and 31%). The HIV-1 RNA results are presented in Table 13.
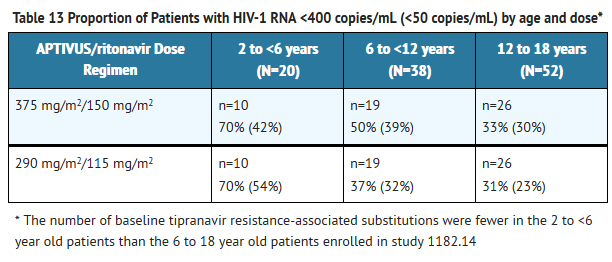
This image is provided by the National Library of Medicine.

- The dose selection for all age groups was based on the following:
- A greater proportion of patients receiving APTIVUS/ritonavir 375 mg/m2/150 mg/m2 compared to 290 mg/m2/115 mg/m2 achieved HIV-1 RNA <400 and <50 copies/mL.
- A greater proportion of patients 6 to 18 years of age with multiple baseline protease inhibitor resistance-associated substitutions receiving APTIVUS/ritonavir 375 mg/m2/150 mg/m2 achieved HIV-1 RNA <400 copies/mL at 48 weeks compared to patients receiving APTIVUS/ritonavir 290 mg/m2/115 mg/m2.
- No clinically significant increase in adverse event rates observed with 375 mg/m2/150 mg/m2 compared to 290 mg/m2/115 mg/m2.
- Overall, 6 (5%) patients ages 6 to 18 had AIDS defining illness during the treatment period and all received the 290 mg/m2/115 mg/m2 dose.
- The guidance for possible dose reduction for patients who develop intolerance or toxicity and cannot continue with APTIVUS/ritonavir 14 mg/kg/6 mg/kg (or 375 mg/m2/150 mg/m2) is based on the following:
- The 290 mg/m2/115 mg/m2 twice daily regimen provided tipranavir plasma concentrations similar to those obtained in adults receiving 500/200 mg twice daily. The 375 mg/m2/150 mg/m2 twice daily regimen provided tipranavir plasma concentrations 37% higher than those obtained in adults receiving 500/200 mg twice daily.
- The observed response rates for APTIVUS/ritonavir dose of 290 mg/m2/115 mg/m2 as shown in Table 13.
- Dose reduction is not appropriate for patients whose virus is resistant to more than one protease inhibitor.
- When body surface area (BSA) dosing is converted to mg/kg dosing, the APTIVUS/ritonavir 375 mg/m2/150 mg/m2 twice daily regimen is similar to 14 mg/kg/6 mg/kg and APTIVUS/ritonavir 290 mg/m2/115 mg/m2 twice daily regimen is similar to 12 mg/kg/5 mg/kg twice daily.
How Supplied
- APTIVUS capsules 250 mg are pink, oblong soft gelatin capsules imprinted in black with "TPV 250". They are packaged in HDPE unit-of-use bottles with a child resistant closure and 120 capsules. (NDC 0597-0003-02)
- APTIVUS oral solution is a clear yellow viscous buttermint-butter toffee flavored liquid containing 100 mg tipranavir in each mL. The solution is supplied in a unit-of-use amber glass bottle providing 95 mL of solution with a child resistant closure. A 5 mL plastic oral dispensing syringe is also provided. (NDC 0597-0002-01).
- Storage
- APTIVUS capsules should be stored in a refrigerator 2°-8°C (36°-46°F) prior to opening the bottle. After opening the bottle, the capsules may be stored at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F) and must be used within 60 days after first opening the bottle.
- APTIVUS oral solution should be stored at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F). Do not refrigerate or freeze. The solution must be used within 60 days after first opening the bottle.
- Store in a safe place out of the reach of children.
Storage
There is limited information regarding Tipranavir Storage in the drug label.
Images
Drug Images
 |
Drug Name: |
| This pill image is provided by the National Library of Medicine's PillBox. |
Package and Label Display Panel
 |
| This image of the FDA label is provided by the National Library of Medicine. |
 |
| This image of the FDA label is provided by the National Library of Medicine. |
 |
| This image of the FDA label is provided by the National Library of Medicine. |
 |
| This image of the FDA label is provided by the National Library of Medicine. |
Patient Counseling Information
- Hepatic Impairment and Toxicity
- Inform patients that APTIVUS co-administered with 200 mg of ritonavir, has been associated with severe liver disease, including some deaths. Patients with signs or symptoms of clinical hepatitis should discontinue APTIVUS/ritonavir treatment and seek medical evaluation. Symptoms of hepatitis include fatigue, malaise, anorexia, nausea, jaundice, bilirubinuria, acholic stools, liver tenderness or hepatomegaly. Extra vigilance is needed for patients with chronic hepatitis B or C co-infection, as these patients have an increased risk of developing hepatotoxicity.
- Liver function tests should be performed prior to initiating therapy with APTIVUS and 200 mg of ritonavir, and frequently throughout the duration of treatment. Patients with chronic hepatitis B or C co-infection or elevations in liver enzymes prior to treatment are at increased risk (approximately 2-fold) for developing further liver enzyme elevations or severe liver disease. Caution should be exercised when administering APTIVUS/ritonavir to patients with liver enzyme abnormalities or history of chronic liver disease. Increased liver function testing is warranted in these patients. APTIVUS should not be given to patients with moderate to severe hepatic impairment.
- Intracranial Hemorrhage
- Inform patients that APTIVUS co-administered with 200 mg of ritonavir has been associated with reports of both fatal and non-fatal intracranial hemorrhage. Patients should report any unusual or unexplained bleeding to their physician.
- Drug Interactions
- APTIVUS may interact with some drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or non-prescription medications or herbal products, particularly St. John’s wort.
- Use of Vitamin E
- Advise patients taking APTIVUS oral solution not to take supplemental vitamin E greater than a standard multivitamin as APTIVUS oral solution contains 116 IU/mL of vitamin E and when taken at the recommended maximum dose of 500 mg/200 mg tipranavir/ritonavir BID, results in a daily dose of 1160 IU. This intake is higher than the Reference Daily Intake (adults 30 IU, pediatrics approximately 10 IU).
- Rash
- Rash, including flat or raised rashes or sensitivity to the sun, have been reported in approximately 10% of subjects receiving APTIVUS. Some patients who developed rash also had one or more of the following symptoms: joint pain or stiffness, throat tightness, generalized itching, muscle aches, fever, redness, blisters, or peeling of the skin. Women taking birth control pills may get a skin rash. Tell patients to discontinue use of APTIVUS and call their physician right away if any of these symptoms develop.
- Sulfa Allergy
- Tell patients to report any history of sulfonamide allergy to the physician.
- Contraceptives
- Women receiving estrogen-based hormonal contraceptives should be instructed that additional or alternative contraceptive measures should be used during therapy with APTIVUS/ritonavir. There may be an increased risk of rash when APTIVUS is given with hormonal contraceptives.
- Fat Redistribution
- Inform patients that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time.
- Administration
- Inform patients that APTIVUS must be co-administered with ritonavir to ensure its therapeutic effect. Failure to correctly co-administer APTIVUS with ritonavir will result in reduced plasma levels of tipranavir that may be insufficient to achieve the desired antiviral effect.
- APTIVUS co-administered with ritonavir capsules or solution can be taken with or without meals
- APTIVUS co-administered with ritonavir tablets must only be taken with meals
- Tell patients that sustained decreases in plasma HIV-1 RNA have been associated with a reduced risk of progression to AIDS and death. Patients should remain under the care of a physician while using APTIVUS. Advise patients to take APTIVUS and other concomitant antiretroviral therapy every day as prescribed. APTIVUS, co-administered with ritonavir, must be given in combination with other antiretroviral drugs. Patients should not alter the dose or discontinue therapy without consulting with their healthcare professional. If a dose of APTIVUS is missed, patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped the patient should not double the next dose.
- APTIVUS is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using APTIVUS.
- Patients should be advised to avoid doing things that can spread HIV-1 infection to others.
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
- Do not breastfeed. It is not known if APTIVUS can be passed to your baby in your breast milk and whether it could harm your baby. Also, mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.

This image is provided by the National Library of Medicine.

Precautions with Alcohol
- Alcohol-Tipranavir interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- APTIVUS®[1]
Look-Alike Drug Names
There is limited information regarding Tipranavir Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.