Pheochromocytoma pathophysiology: Difference between revisions
No edit summary |
No edit summary |
||
| Line 23: | Line 23: | ||
* 60-65 percent of pheochromocytomas are sporadic.<ref name="pmid6103678">{{cite journal |vauthors=Webb TA, Sheps SG, Carney JA |title=Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2 |journal=Am. J. Surg. Pathol. |volume=4 |issue=2 |pages=121–6 |year=1980 |pmid=6103678 |doi= |url=}}</ref><ref name="pmid3474647">{{cite journal |vauthors=Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T |title=Gene expression from transcriptionally disabled retroviral vectors |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=84 |issue=15 |pages=5197–201 |year=1987 |pmid=3474647 |pmc=298821 |doi= |url=}}</ref> | * 60-65 percent of pheochromocytomas are sporadic.<ref name="pmid6103678">{{cite journal |vauthors=Webb TA, Sheps SG, Carney JA |title=Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2 |journal=Am. J. Surg. Pathol. |volume=4 |issue=2 |pages=121–6 |year=1980 |pmid=6103678 |doi= |url=}}</ref><ref name="pmid3474647">{{cite journal |vauthors=Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T |title=Gene expression from transcriptionally disabled retroviral vectors |journal=Proc. Natl. Acad. Sci. U.S.A. |volume=84 |issue=15 |pages=5197–201 |year=1987 |pmid=3474647 |pmc=298821 |doi= |url=}}</ref> | ||
* Pheochromocytomas can be familial and occur in patients with [[multiple endocrine neoplasia]]<nowiki/> | * Pheochromocytomas can be familial and occur in patients with [[multiple endocrine neoplasia]]<nowiki/> ([[Multiple endocrine neoplasia type 1|MEN1]] and [[Multiple endocrine neoplasia type 2|MEN 2B]]). | ||
* Patients with Von Hippel Lindau disease ([[VHL]]) may also develop pheochromocytoma.<ref name="pmid24642075">{{cite journal| author=Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM| title=The genetic basis of pheochromocytoma and paraganglioma: implications for management. | journal=Urology | year= 2014 | volume= 83 | issue= 6 | pages= 1225-32 | pmid=24642075 | doi=10.1016/j.urology.2014.01.007 | pmc=4572836 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=24642075 }}</ref> | * Patients with [[Von Hippel-Lindau disease|Von Hippel Lindau disease]] ([[VHL]]) may also develop pheochromocytoma.<ref name="pmid24642075">{{cite journal| author=Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM| title=The genetic basis of pheochromocytoma and paraganglioma: implications for management. | journal=Urology | year= 2014 | volume= 83 | issue= 6 | pages= 1225-32 | pmid=24642075 | doi=10.1016/j.urology.2014.01.007 | pmc=4572836 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=24642075 }}</ref> | ||
* It has [[Autosomal dominant inheritance|autosomal dominant]] inheritance and has two pathways of [[tumor]] pathogenesis. Cluster 1 tumors are [[noradrenergic]]. Cluster 2 tumors are [[adrenergic]].<ref name="pmid23933153">{{cite journal| author=King KS, Pacak K| title=Familial pheochromocytomas and paragangliomas. | journal=Mol Cell Endocrinol | year= 2014 | volume= 386 | issue= 1-2 | pages= 92-100 | pmid=23933153 | doi=10.1016/j.mce.2013.07.032 | pmc=3917973 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=23933153 }}</ref> | * It has [[Autosomal dominant inheritance|autosomal dominant]] inheritance and has two pathways of [[tumor]] pathogenesis. Cluster 1 tumors are [[noradrenergic]]. Cluster 2 tumors are [[adrenergic]].<ref name="pmid23933153">{{cite journal| author=King KS, Pacak K| title=Familial pheochromocytomas and paragangliomas. | journal=Mol Cell Endocrinol | year= 2014 | volume= 386 | issue= 1-2 | pages= 92-100 | pmid=23933153 | doi=10.1016/j.mce.2013.07.032 | pmc=3917973 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=23933153 }}</ref> | ||
{| class="wikitable" | {| class="wikitable" | ||
! colspan="2" |Familial | ! colspan="2" |Familial pheocromocytoma | ||
|- | |- | ||
!Cluster 1 (Noradrenergic) | !Cluster 1 (Noradrenergic) | ||
| Line 41: | Line 41: | ||
* '''[[Neurofibromatosis type I|Neurofibromatosis type 1]] (NF1)''' | * '''[[Neurofibromatosis type I|Neurofibromatosis type 1]] (NF1)''' | ||
|} | |} | ||
* Patients with the [[Succinate dehydrogenase|succinate dehydrogenase B]] mutations are likely to develop a malignant disease.<ref name="pmid15328326">{{cite journal| author=Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M et al.| title=Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. | journal=JAMA | year= 2004 | volume= 292 | issue= 8 | pages= 943-51 | pmid=15328326 | doi=10.1001/jama.292.8.943 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=15328326 }}</ref> | * Patients with the [[Succinate dehydrogenase|succinate dehydrogenase B]] [[mutations]] are likely to develop a [[malignant]] disease.<ref name="pmid15328326">{{cite journal| author=Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M et al.| title=Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. | journal=JAMA | year= 2004 | volume= 292 | issue= 8 | pages= 943-51 | pmid=15328326 | doi=10.1001/jama.292.8.943 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=15328326 }}</ref> | ||
* [[Von Hippel-Lindau Disease|Von Hippel-Lindau]] (VHL) disease | * [[Von Hippel-Lindau Disease|Von Hippel-Lindau]] ([[Von Hippel-Lindau tumor suppressor|VHL]]) disease | ||
**PCCs arise in about 10–20% of patients with VHL disease. | **PCCs arise in about 10–20% of patients with [[VHL]] disease. | ||
**VHL tumor suppressor protein is the main cause of the disease. | **VHL [[Tumor suppressor gene|tumor suppressor protein]] is the main cause of the disease.tcted. | ||
**The VHL tumor suppressor protein targets especially hypoxia-inducible factor-1 (HIF-1), MMP inhibitors, and atypical protein kinase C.<ref name="pmid12209156">{{cite journal| author=Kaelin WG| title=Molecular basis of the VHL hereditary cancer syndrome. | journal=Nat Rev Cancer | year= 2002 | volume= 2 | issue= 9 | pages= 673-82 | pmid=12209156 | doi=10.1038/nrc885 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=12209156 }}</ref> | **The [[VHL]] [[Tumor suppressor gene|tumor suppressor protein]] targets especially hypoxia-inducible factor-1 (HIF-1), [[MMP]] inhibitors, and atypical [[protein kinase C]].<ref name="pmid12209156">{{cite journal| author=Kaelin WG| title=Molecular basis of the VHL hereditary cancer syndrome. | journal=Nat Rev Cancer | year= 2002 | volume= 2 | issue= 9 | pages= 673-82 | pmid=12209156 | doi=10.1038/nrc885 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=12209156 }}</ref> | ||
**HIF-1 is involved in erythropoiesis through its ability to induce transcription of mRNA coding for erythropoietin. it regulates several growth factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF)-beta, and transforming growth factor (TGF | **HIF-1 is involved in [[erythropoiesis]] through its ability to induce [[Transcription (genetics)|transcription]] of [[mRNA]] coding for [[erythropoietin]]. it regulates several [[growth factors]], such as [[vascular endothelial growth factor]] (VEGF), [[platelet-derived growth factor]] (PDGF)-beta, and [[transforming growth factor]] (TGF-alpha).<ref name="pmid15350900">{{cite journal| author=Barry RE, Krek W| title=The von Hippel-Lindau tumour suppressor: a multi-faceted inhibitor of tumourigenesis. | journal=Trends Mol Med | year= 2004 | volume= 10 | issue= 9 | pages= 466-72 | pmid=15350900 | doi=10.1016/j.molmed.2004.07.008 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=15350900 }}</ref> | ||
**Deletions in ''VHL'' and nonsense and frameshift mutations appear to be more common in type 1 disease, while missense mutations may be more common in type 2 disease.<ref name="pmid9681856">{{cite journal| author=Neumann HP, Bender BU| title=Genotype-phenotype correlations in von Hippel-Lindau disease. | journal=J Intern Med | year= 1998 | volume= 243 | issue= 6 | pages= 541-5 | pmid=9681856 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=9681856 }}</ref> | **[[Deletion (genetics)|Deletions]] in ''[[VHL]]'' and [[Nonsense mutation|nonsense]] and [[Frameshift mutation|frameshift mutations]] appear to be more common in type 1 disease, while [[missense mutations]] may be more common in type 2 disease.<ref name="pmid9681856">{{cite journal| author=Neumann HP, Bender BU| title=Genotype-phenotype correlations in von Hippel-Lindau disease. | journal=J Intern Med | year= 1998 | volume= 243 | issue= 6 | pages= 541-5 | pmid=9681856 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=9681856 }}</ref> | ||
**[[Missense mutations]] at codon 167 are associated with a particularly high risk of PCC.<ref name="pmid8730290">{{cite journal| author=Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ et al.| title=Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. | journal=J Med Genet | year= 1996 | volume= 33 | issue= 4 | pages= 328-32 | pmid=8730290 | doi= | pmc=1050584 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=8730290 }}</ref> | |||
*'''[[Multiple endocrine neoplasia, type 2|Multiple endocrine neoplasia]] type 2A''' | *'''[[Multiple endocrine neoplasia, type 2|Multiple endocrine neoplasia]] type 2A''' | ||
**The RET protein is a transmembrane receptor of the tyrosine kinase family. | **The [[RET gene|RET]] protein is a transmembrane [[receptor]] of the [[tyrosine kinase]] family. | ||
**It is | **It is derived from the [[neural crest]] and has a key role in regulating [[cell proliferation]] and survival during [[embryogenesis]].<ref name="pmid7907913">{{cite journal| author=Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E et al.| title=Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. | journal=Nat Genet | year= 1994 | volume= 6 | issue= 1 | pages= 70-4 | pmid=7907913 | doi=10.1038/ng0194-70 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=7907913 }}</ref> | ||
**The RET receptor can be activated through various factors such as glial-cell-line-derived neurotrophic factor (GDNF), neurturin, artemin, and persephin.<ref name="pmid11073534">{{cite journal| author=Hansford JR, Mulligan LM| title=Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. | journal=J Med Genet | year= 2000 | volume= 37 | issue= 11 | pages= 817-27 | pmid=11073534 | doi= | pmc=1734482 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=11073534 }}</ref> | **The [[RET gene|RET]] receptor can be activated through various factors such as [[Glial cell line-derived neurotrophic factor|glial-cell-line-derived neurotrophic factor]] ([[GDNF]]), [[neurturin]], [[artemin]], and [[persephin]].<ref name="pmid11073534">{{cite journal| author=Hansford JR, Mulligan LM| title=Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. | journal=J Med Genet | year= 2000 | volume= 37 | issue= 11 | pages= 817-27 | pmid=11073534 | doi= | pmc=1734482 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=11073534 }}</ref> | ||
** | **[[Mutations]] of the [[RET proto-oncogene|''RET'' proto-oncogene]] cause constitutive activation of the [[RET gene|RET]] receptor and of intracellular signaling pathways, ultimately resulting in the cellular transformation.<ref name="pmid7532281">{{cite journal| author=Asai N, Iwashita T, Matsuyama M, Takahashi M| title=Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. | journal=Mol Cell Biol | year= 1995 | volume= 15 | issue= 3 | pages= 1613-9 | pmid=7532281 | doi= | pmc=230385 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=7532281 }}</ref> | ||
**Mutations causing loss of function of the RET protein were found to be associated with Hirschsprung's disease, a | **[[Mutations]] causing loss of function of the [[RET gene|RET]] protein were found to be associated with [[Hirschsprung's disease]], a disorder characterized by the absence of [[enteric ganglia]] in the [[Gastrointestinal tract|intestinal tract]].<ref name="pmid16448984">{{cite journal| author=Lantieri F, Griseri P, Ceccherini I| title=Molecular mechanisms of RET-induced Hirschsprung pathogenesis. | journal=Ann Med | year= 2006 | volume= 38 | issue= 1 | pages= 11-9 | pmid=16448984 | doi=10.1080/07853890500442758 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=16448984 }}</ref> | ||
* '''[[Neurofibromatosis type I|Neurofibromatosis type 1]] (NF1)''' | * '''[[Neurofibromatosis type I|Neurofibromatosis type 1]] (NF1)''' | ||
**The ''NF1'' gene has been localized on chromosome 17qll.2 and encodes | **[[Mutations]] in the [[NF1|''NF1'' gene]] result in loss of functional [[protein]] causing the wide spectrum of clinical findings. | ||
** | **The ''[[NF1]]'' gene has been localized on [[chromosome]] 17qll.2 and encodes [[neurofibromin]]. In the absence or at decreased levels of [[neurofibromin]], [[Signaling pathway|signaling]] is increased through various pathways resulting in the [[cell proliferation]] and inhibited [[apoptosis]].<ref name="pmid7926784">{{cite journal| author=Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW et al.| title=Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. | journal=Genes Dev | year= 1994 | volume= 8 | issue= 9 | pages= 1019-29 | pmid=7926784 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=7926784 }}</ref><ref name="pmid8825042">{{cite journal| author=Shen MH, Harper PS, Upadhyaya M| title=Molecular genetics of neurofibromatosis type 1 (NF1). | journal=J Med Genet | year= 1996 | volume= 33 | issue= 1 | pages= 2-17 | pmid=8825042 | doi= | pmc=1051805 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=8825042 }}</ref> | ||
**[[Knudson hypothesis|Knudson's two-hit tumor suppressor model]] could be applied, resulting in a [[loss of heterozygosity]] at tumor level. <ref name="pmid7519874">{{cite journal| author=Gutmann DH, Cole JL, Stone WJ, Ponder BA, Collins FS| title=Loss of neurofibromin in adrenal gland tumors from patients with neurofibromatosis type I. | journal=Genes Chromosomes Cancer | year= 1994 | volume= 10 | issue= 1 | pages= 55-8 | pmid=7519874 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=7519874 }}</ref>The [[mutations]] include [[translocations]], [[Splicing (genetics)|splicing]], [[Deletion (genetics)|deletions]], [[insertions]], and [[point mutations]]. | |||
**The Ras-GTPase-activating protein-related domain has the important role of stimulating the intrinsic [[GTPase]] of p21-Ras''-''GTP to hydrolyze GTP to GDP and inactivating p21-Ras. .[[P21|P21-Ras]] is a key component of many [[growth factors]] signaling pathways, and [[neurofibromin]] acts as a [[Tumor suppressor|tumor suppressor protein]]. | |||
**The | **The cysteine-serine-rich domain ([[CSR]]) of [[neurofibromin]] plays an important role in the pathogenesis of NF1.<ref name="pmid17426081">{{cite journal| author=Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M et al.| title=Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. | journal=J Clin Endocrinol Metab | year= 2007 | volume= 92 | issue= 7 | pages= 2784-92 | pmid=17426081 | doi=10.1210/jc.2006-2833 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=17426081 }}</ref> | ||
** | |||
==Associated conditions== | ==Associated conditions== | ||
* Pheochromocytoma can be part of other syndromes named [[Multiple endocrine neoplasia|multiple endocrine | * Pheochromocytoma can be part of other syndromes named [[Multiple endocrine neoplasia|multiple endocrine neoplasia]] ([[Multiple endocrine neoplasia type 1|MEN1]] and [[MEN2|MEN2B]]), which are [[Autosomal dominant inheritance|autosomal dominant]] syndromes controlled by [[RET proto-oncogene|RET gene]]. Pheochromocytoma occurs in 50% of patients with [[MEN2]] as follows: | ||
{| class="wikitable" | {| class="wikitable" | ||
!MEN1 | !MEN1 | ||
| Line 87: | Line 85: | ||
==Gross Pathology== | ==Gross Pathology== | ||
*On [[gross pathology]], pheochromocytoma varies from small to large and usually associated with [[hemorrhage]] and [[necrosis]].<ref name="pmid26266130">{{cite journal| author=Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB| title=Non Functional Unilateral Adrenal Myelolipoma, A Case Report. | journal=J Clin Diagn Res | year= 2015 | volume= 9 | issue= 6 | pages= ED03-4 | pmid=26266130 | doi=10.7860/JCDR/2015/13209.6070 | pmc=4525519 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=26266130 }}</ref> | *On [[gross pathology]], pheochromocytoma varies from small to large and usually associated with [[hemorrhage]] and [[necrosis]].<ref name="pmid26266130">{{cite journal| author=Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB| title=Non Functional Unilateral Adrenal Myelolipoma, A Case Report. | journal=J Clin Diagn Res | year= 2015 | volume= 9 | issue= 6 | pages= ED03-4 | pmid=26266130 | doi=10.7860/JCDR/2015/13209.6070 | pmc=4525519 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=26266130 }}</ref> | ||
*Pheochromocytoma | *Pheochromocytoma is usually lobulated and small [[tumors]] have compressed [[adrenal gland]]. | ||
*Familial [[Tumor|tumors]] are bilateral. | *Familial [[Tumor|tumors]] are bilateral. | ||
*It may be associated with hyperplasia in the adjacent [[medulla]]. | *It may be associated with hyperplasia in the adjacent [[medulla]]. | ||
*[[Chromaffin]] reaction: fresh [[tumor]] cut section turns dark brown if add [[potassium dichromate]] at pH 5-6. | *[[Chromaffin]] reaction: fresh [[tumor]] cut section turns dark brown if add [[potassium dichromate]] at pH 5-6. | ||
<gallery> | <gallery> | ||
Image:Bilateral pheo MEN2.jpg|Bilateral pheochromocytoma in [[Multiple_endocrine_neoplasia_type_2|MEN2]]. Gross image. | Image:Bilateral pheo MEN2.jpg|Bilateral pheochromocytoma in [[Multiple_endocrine_neoplasia_type_2|MEN2]]. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg | ||
</gallery> | </gallery> | ||
| Line 98: | Line 96: | ||
On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma. | On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma. | ||
<gallery> | <gallery> | ||
Image:Adrenal pheochromocytoma (1) histopathology.jpg|[[Micrograph]] of pheochromocytoma. | Image:Adrenal pheochromocytoma (1) histopathology.jpg|[[Micrograph]] of pheochromocytoma. Source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524 | ||
Image:Adrenal pheochromocytoma (3) histopathology.jpg|Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. | Image:Adrenal pheochromocytoma (3) histopathology.jpg|Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945 | ||
Image:Adrenal pheochromocytoma (2) histopathology.jpg|Micrograph of pheochromocytoma. | Image:Adrenal pheochromocytoma (2) histopathology.jpg|Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944 | ||
Image:Adrenal pheochromocytoma (3) histopathology.jpg|Micrograph of pheochromocytoma. | Image:Adrenal pheochromocytoma (3) histopathology.jpg|Micrograph of pheochromocytoma. | ||
</gallery> | </gallery> | ||
Revision as of 14:19, 3 October 2017
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma pathophysiology |
|
Risk calculators and risk factors for Pheochromocytoma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytoma arises from chromaffin cells of the adrenal medulla. On gross pathology, pheochromocytoma has a multinodular and a multicentric pattern of growth. On microscopic histopathological analysis, nesting (Zellballen) pattern composed of well-defined clusters of tumor cells separated by fibrovascular stroma may be seen. It may be benign, malignant, familial(multiple endocrine neoplasia 1 and type 2B) or sporadic. All of these forms have genetic origin depending on a large number of genes, for example, VHL, SDH, NF1, RET genes.
Pathophysiology
- Pheochromocytoma arises from chromaffin cells of the adrenal medulla and sympathetic ganglia. Malignant and benign pheochromocytomas share the same biochemical and histological features, the only difference is to have a distant spread or be locally invasive. [1]
Basic physiology of catecholamines[2][3]
- Epinephrine acts on nearly all body tissues. Its actions vary by tissue type and tissue expression of adrenergic receptors.
- Epinephrine is a nonselective agonist of all adrenergic receptors, including the major subtypes α1, α2, β1, β2, and β3:
- Binding to α1 receptors causes vasoconstriction. Blood vessels with α1-adrenergic receptors are present in the skin, the sphincters of the gastrointestinal system, kidney (renal artery) and brain. During the fight-or-flight response vasoconstriction results in decreased blood flow to these organs.
- Binding to α2 receptors inhibits insulin secretion by the pancreas, stimulates glycogenolysis in the liver and muscle, and stimulates glycolysis and inhibits insulin-mediated glycogenesis in muscle. It suppresses the release of norepinephrine by negative feedback.
- Binding to β2 receptors causes Smooth muscle relaxation in the uterus, GI tract, detrusor urinae muscle of bladder wall, and bronchi. It also causes dilatation of smaller coronary arteries, hepatic artery, arteries to skeletal muscle.
- Binding to β1 receptors causes renin release from juxtaglomerular cells and lipolysis in adipose tissue. It Increases cardiac output by:
- Increase in heart rate in sinoatrial node
- Increase in atrial cardiac muscle contractility
- Increases in contractility and automaticity of ventricular cardiac muscle
- Increases in conduction and automaticity of atrioventricular node
Genetics
- Pheochromocytomas can be familial and occur in patients with multiple endocrine neoplasia (MEN1 and MEN 2B).
- Patients with Von Hippel Lindau disease (VHL) may also develop pheochromocytoma.[6]
- It has autosomal dominant inheritance and has two pathways of tumor pathogenesis. Cluster 1 tumors are noradrenergic. Cluster 2 tumors are adrenergic.[7]
| Familial pheocromocytoma | |
|---|---|
| Cluster 1 (Noradrenergic) | Cluster 2 (Adrenergic) |
|
|
- Patients with the succinate dehydrogenase B mutations are likely to develop a malignant disease.[8]
- Von Hippel-Lindau (VHL) disease
- PCCs arise in about 10–20% of patients with VHL disease.
- VHL tumor suppressor protein is the main cause of the disease.tcted.
- The VHL tumor suppressor protein targets especially hypoxia-inducible factor-1 (HIF-1), MMP inhibitors, and atypical protein kinase C.[9]
- HIF-1 is involved in erythropoiesis through its ability to induce transcription of mRNA coding for erythropoietin. it regulates several growth factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF)-beta, and transforming growth factor (TGF-alpha).[10]
- Deletions in VHL and nonsense and frameshift mutations appear to be more common in type 1 disease, while missense mutations may be more common in type 2 disease.[11]
- Missense mutations at codon 167 are associated with a particularly high risk of PCC.[12]
- Multiple endocrine neoplasia type 2A
- The RET protein is a transmembrane receptor of the tyrosine kinase family.
- It is derived from the neural crest and has a key role in regulating cell proliferation and survival during embryogenesis.[13]
- The RET receptor can be activated through various factors such as glial-cell-line-derived neurotrophic factor (GDNF), neurturin, artemin, and persephin.[14]
- Mutations of the RET proto-oncogene cause constitutive activation of the RET receptor and of intracellular signaling pathways, ultimately resulting in the cellular transformation.[15]
- Mutations causing loss of function of the RET protein were found to be associated with Hirschsprung's disease, a disorder characterized by the absence of enteric ganglia in the intestinal tract.[16]
- Neurofibromatosis type 1 (NF1)
- Mutations in the NF1 gene result in loss of functional protein causing the wide spectrum of clinical findings.
- The NF1 gene has been localized on chromosome 17qll.2 and encodes neurofibromin. In the absence or at decreased levels of neurofibromin, signaling is increased through various pathways resulting in the cell proliferation and inhibited apoptosis.[17][18]
- Knudson's two-hit tumor suppressor model could be applied, resulting in a loss of heterozygosity at tumor level. [19]The mutations include translocations, splicing, deletions, insertions, and point mutations.
- The Ras-GTPase-activating protein-related domain has the important role of stimulating the intrinsic GTPase of p21-Ras-GTP to hydrolyze GTP to GDP and inactivating p21-Ras. .P21-Ras is a key component of many growth factors signaling pathways, and neurofibromin acts as a tumor suppressor protein.
- The cysteine-serine-rich domain (CSR) of neurofibromin plays an important role in the pathogenesis of NF1.[20]
Associated conditions
- Pheochromocytoma can be part of other syndromes named multiple endocrine neoplasia (MEN1 and MEN2B), which are autosomal dominant syndromes controlled by RET gene. Pheochromocytoma occurs in 50% of patients with MEN2 as follows:
| MEN1 | MEN2 |
|---|---|
Gross Pathology
- On gross pathology, pheochromocytoma varies from small to large and usually associated with hemorrhage and necrosis.[21]
- Pheochromocytoma is usually lobulated and small tumors have compressed adrenal gland.
- Familial tumors are bilateral.
- It may be associated with hyperplasia in the adjacent medulla.
- Chromaffin reaction: fresh tumor cut section turns dark brown if add potassium dichromate at pH 5-6.
-
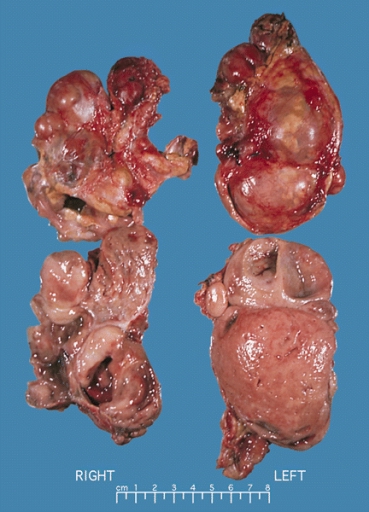
Bilateral pheochromocytoma in MEN2. Gross image. Source: https://upload.wikimedia.org/wikipedia/commons/5/5f/Bilateral_pheo_MEN2.jpg

Microscopic Pathology
On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma.
-
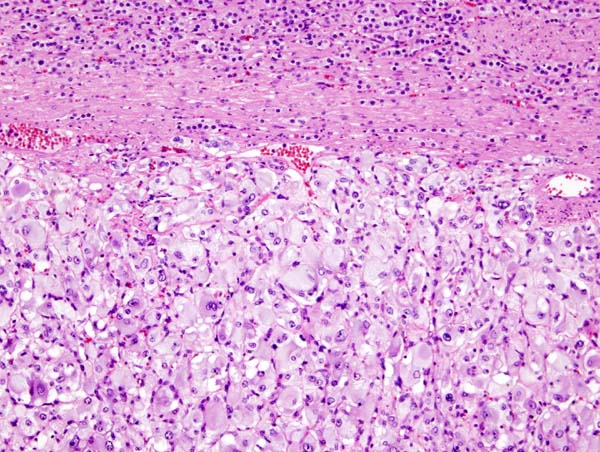
Micrograph of pheochromocytoma. Source: By Nephron - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=5938524
-
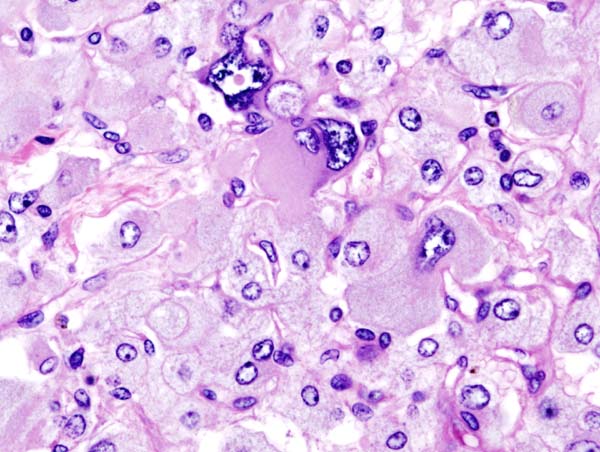
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535945
-
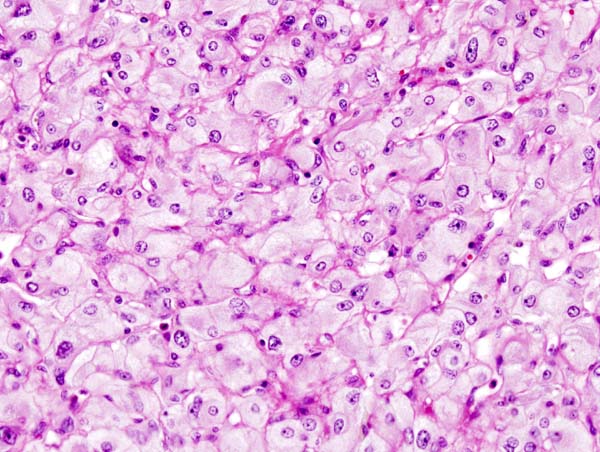
Micrograph of pheochromocytoma. Source: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=535944
-
Micrograph of pheochromocytoma.
_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
{kind=link}
Videos
{{#ev:youtube|7yjxG3KmX98}}
References
- ↑ Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA; et al. (1999). "Clinical experience over 48 years with pheochromocytoma". Ann Surg. 229 (6): 755–64, discussion 764-6. PMC 1420821. PMID 10363888.
- ↑ Raz I, Katz A, Spencer MK (1991). "Epinephrine inhibits insulin-mediated glycogenesis but enhances glycolysis in human skeletal muscle". Am J Physiol. 260 (3 Pt 1): E430–5. PMID 1900669.
- ↑ Arnall DA, Marker JC, Conlee RK, Winder WW (1986). "Effect of infusing epinephrine on liver and muscle glycogenolysis during exercise in rats". Am J Physiol. 250 (6 Pt 1): E641–9. PMID 3521311.
- ↑ Webb TA, Sheps SG, Carney JA (1980). "Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2". Am. J. Surg. Pathol. 4 (2): 121–6. PMID 6103678.
- ↑ Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T (1987). "Gene expression from transcriptionally disabled retroviral vectors". Proc. Natl. Acad. Sci. U.S.A. 84 (15): 5197–201. PMC 298821. PMID 3474647.
- ↑ Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM (2014). "The genetic basis of pheochromocytoma and paraganglioma: implications for management". Urology. 83 (6): 1225–32. doi:10.1016/j.urology.2014.01.007. PMC 4572836. PMID 24642075.
- ↑ King KS, Pacak K (2014). "Familial pheochromocytomas and paragangliomas". Mol Cell Endocrinol. 386 (1–2): 92–100. doi:10.1016/j.mce.2013.07.032. PMC 3917973. PMID 23933153.
- ↑ Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M; et al. (2004). "Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations". JAMA. 292 (8): 943–51. doi:10.1001/jama.292.8.943. PMID 15328326.
- ↑ Kaelin WG (2002). "Molecular basis of the VHL hereditary cancer syndrome". Nat Rev Cancer. 2 (9): 673–82. doi:10.1038/nrc885. PMID 12209156.
- ↑ Barry RE, Krek W (2004). "The von Hippel-Lindau tumour suppressor: a multi-faceted inhibitor of tumourigenesis". Trends Mol Med. 10 (9): 466–72. doi:10.1016/j.molmed.2004.07.008. PMID 15350900.
- ↑ Neumann HP, Bender BU (1998). "Genotype-phenotype correlations in von Hippel-Lindau disease". J Intern Med. 243 (6): 541–5. PMID 9681856.
- ↑ Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ; et al. (1996). "Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations". J Med Genet. 33 (4): 328–32. PMC 1050584. PMID 8730290.
- ↑ Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E; et al. (1994). "Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC". Nat Genet. 6 (1): 70–4. doi:10.1038/ng0194-70. PMID 7907913.
- ↑ Hansford JR, Mulligan LM (2000). "Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis". J Med Genet. 37 (11): 817–27. PMC 1734482. PMID 11073534.
- ↑ Asai N, Iwashita T, Matsuyama M, Takahashi M (1995). "Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations". Mol Cell Biol. 15 (3): 1613–9. PMC 230385. PMID 7532281.
- ↑ Lantieri F, Griseri P, Ceccherini I (2006). "Molecular mechanisms of RET-induced Hirschsprung pathogenesis". Ann Med. 38 (1): 11–9. doi:10.1080/07853890500442758. PMID 16448984.
- ↑ Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW; et al. (1994). "Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues". Genes Dev. 8 (9): 1019–29. PMID 7926784.
- ↑ Shen MH, Harper PS, Upadhyaya M (1996). "Molecular genetics of neurofibromatosis type 1 (NF1)". J Med Genet. 33 (1): 2–17. PMC 1051805. PMID 8825042.
- ↑ Gutmann DH, Cole JL, Stone WJ, Ponder BA, Collins FS (1994). "Loss of neurofibromin in adrenal gland tumors from patients with neurofibromatosis type I." Genes Chromosomes Cancer. 10 (1): 55–8. PMID 7519874.
- ↑ Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M; et al. (2007). "Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1". J Clin Endocrinol Metab. 92 (7): 2784–92. doi:10.1210/jc.2006-2833. PMID 17426081.
- ↑ Sajjanar AB, Athanikar VS, Dinesh US, Nanjappa B, Patil PB (2015). "Non Functional Unilateral Adrenal Myelolipoma, A Case Report". J Clin Diagn Res. 9 (6): ED03–4. doi:10.7860/JCDR/2015/13209.6070. PMC 4525519. PMID 26266130.