Telbivudine
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vignesh Ponnusamy, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
WARNING
See full prescribing information for complete Boxed Warning.
LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS AND SEVERE ACUTE EXACERBATIONS OF HEPATITIS B:
|
Overview
Telbivudine is an HBV nucleoside analogue reverse transcriptase inhibitor that is FDA approved for the treatment of chronic hepatitis B in adult patients with evidence of viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease. There is a Black Box Warning for this drug as shown here. Common adverse reactions include fatigue, increased creatine kinase (CK), headache, cough, diarrhea, abdominal pain, nausea, pharyngolaryngeal pain, arthralgia, pyrexia, rash, back pain, dizziness, myalgia, ALT increased, dyspepsia, insomnia, and abdominal distension.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Chronic Hepatitis B
- Due to higher rates of resistance that may develop with longer term treatment among patients with incomplete viral suppression, treatment should only be initiated, if pre-treatment HBV DNA and ALT measurements are known, in the following patient populations:
- For HBeAg-positive patients, HBV DNA should be less than 9 log10 copies per mL and ALT should be greater than or equal to 2x ULN prior to treatment with Tyzeka.
- For HBeAg-negative patients, HBV DNA should be less than 7 log10 copies per mL prior to treatment with Tyzeka.
- HBV DNA levels should be monitored at 24 weeks of treatment to assure complete viral suppression (HBV DNA less than 300 copies per mL). Alternate therapy should be initiated for patients who have detectable HBV DNA after 24 weeks of treatment. Optimal therapy should be guided by further resistance testing.
- The recommended dose of Tyzeka for the treatment of chronic hepatitis B is 600 mg once daily, taken orally, with or without food.
- Tyzeka oral solution (30 mL) may be considered for patients who have difficulty with swallowing tablets.
- Duration of Therapy
- For patients with incomplete viral suppression (HBV DNA greater than or equal to 300 copies per mL) after 24 weeks of treatment, alternate therapy should be instituted. HBV DNA should be monitored every 6 months to assure continued response. If patients test positive for HBV DNA at any time after their initial response, alternate treatment should be instituted. Optimal therapy should be guided by resistance testing.
- The optimal duration of therapy with Tyzeka for patients with chronic hepatitis B is unknown.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Telbivudine in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Telbivudine in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Telbivudine in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Telbivudine in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Telbivudine in pediatric patients.
Contraindications
- Combination of Tyzeka with pegylated interferon alfa-2a is contraindicated because of increased risk of peripheral neuropathy.
Warnings
|
WARNING
See full prescribing information for complete Boxed Warning.
LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS AND SEVERE ACUTE EXACERBATIONS OF HEPATITIS B:
|
Precautions
- Lactic Acidosis
- Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination with antiretrovirals. Female gender, obesity, and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering HBV nucleoside analogue reverse transcriptase inhibitors to patients with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with Tyzeka should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
- Exacerbations of Hepatitis B after Discontinuation of Treatment
- Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy, including Tyzeka. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, resumption of anti-hepatitis B therapy may be warranted.
- Myopathy
- Cases of myopathy/myositis have been reported with Tyzeka use several weeks to months after starting therapy. Myopathy has also been reported with some other drugs in this class. Rhabdomyolysis has been reported during postmarketing use of Tyzeka.
- Uncomplicated myalgia has been reported in Tyzeka-treated patients. Myopathy, defined as persistent unexplained muscle aches and/or muscle weakness in conjunction with increases in creatine kinase (CK) values, should be considered in any patient with diffuse myalgias, muscle tenderness, or muscle weakness. Among patients with Tyzeka-associated myopathy, no pattern with regard to the degree or timing of CK elevations has been observed. In addition, the predisposing factors for the development of myopathy among Tyzeka recipients are unknown. Patients should be advised to report promptly unexplained muscle aches, pain, tenderness, or weakness. Tyzeka therapy should be interrupted if myopathy is suspected, and discontinued if myopathy is confirmed. It is unknown whether the risk of myopathy during treatment with drugs in this class is increased with concurrent administration of other drugs associated with myopathy, including but not limited to: corticosteroids, chloroquine, hydroxychloroquine, certain HMGCoA reductase inhibitors, fibric acid derivatives, penicillamine, zidovudine, cyclosporine, erythromycin, niacin, and certain azole antifungals. Physicians initiating concomitant treatment with any drug associated with myopathy should monitor patients closely for any signs or symptoms of unexplained muscle pain, tenderness, or weakness.
- Peripheral Neuropathy
- Peripheral neuropathy has been reported with Tyzeka alone or in combination with pegylated interferon alfa-2a and other interferons. In one clinical trial, an increased risk and severity of peripheral neuropathy was observed with the combination use of Tyzeka 600mg daily and pegylated interferon alfa-2a 180 micrograms once weekly compared to Tyzeka or pegylated interferon alfa-2a alone. Such risk cannot be excluded for other dose regimens of pegylated interferon alfa-2a, or other alfa interferons (pegylated or standard). The safety and efficacy of Tyzeka in combination with pegylated interferons or other interferons for the treatment of chronic hepatitis B have not been demonstrated. Patients should be advised to report any numbness, tingling, and/or burning sensations in the arms and/or legs, with or without gait disturbance. Tyzeka therapy should be interrupted if peripheral neuropathy is suspected, and discontinued if peripheral neuropathy is confirmed.
Adverse Reactions
Clinical Trials Experience
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
- Assessment of adverse reactions is primarily based on two trials (007 GLOBE and NV-02B-015) in which 1,699 subjects with chronic hepatitis B received double-blind treatment with Tyzeka 600 mg per day (n=847 subjects) or lamivudine (n=852 subjects) for 104 weeks. The median duration of therapy was 104 weeks for both treatment groups.
- In the 104 week clinical trials, most adverse experiences reported with Tyzeka were classified as mild or moderate in severity and were not attributed to Tyzeka. Selected adverse events of any severity which were reported in greater than or equal to 3% of Tyzeka and lamivudine recipients are shown in Table 2. With the exception of increased creatine kinase (CK), which was reported more frequently among Tyzeka recipients, the adverse event profile was similar for the two drugs.
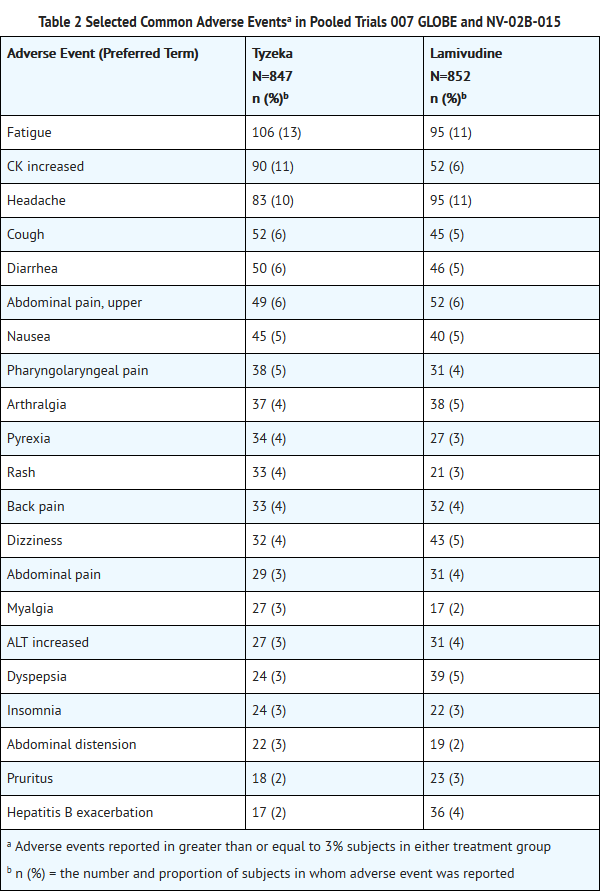
This image is provided by the National Library of Medicine.

- Moderate to severe (Grade 2-4) adverse events were reported in 239/847 (28%) of Tyzeka recipients and 229/852 (27%) of lamivudine recipients. The profile of adverse events of moderate to severe intensity was similar in both treatment groups and no individual adverse event was reported in greater than 2% of subjects in either treatment group.
- Discontinuations due to adverse events were reported in 4% of Tyzeka recipients and 4% of lamivudine recipients. The most common adverse events resulting in Tyzeka discontinuation included increased CK, nausea, diarrhea, fatigue, myalgia, and myopathy.
- Peripheral neuropathy was reported as an adverse event in less than 1% (2/847) of subjects receiving Tyzeka monotherapy. Of Tyzeka-treated subjects less than 1% (5/847) were diagnosed with myopathy/myositis (presenting with muscular weakness).
- Laboratory Abnormalities
- Frequencies of selected treatment-emergent laboratory abnormalities in the 007 GLOBE and NV-02B-015 trials are listed in Table 3.
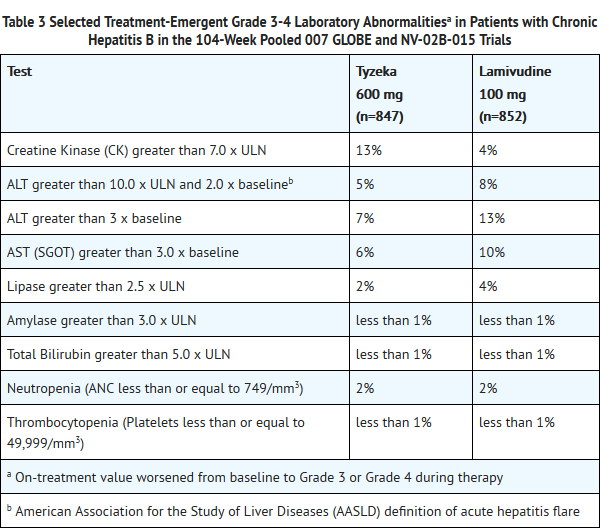
This image is provided by the National Library of Medicine.

- Creatine Kinase (CK) Elevations
- Creatine kinase (CK) elevations were more frequent among subjects on Tyzeka treatment. By 104 weeks of treatment, Grade 1-4 CK elevations occurred in 79% of Tyzeka-treated subjects and 47% of lamivudine-treated subjects. Grade 3 or 4 CK elevations occurred in 13% of Tyzeka-treated subjects and 4% of lamivudine-treated subjects. Most CK elevations were asymptomatic, but the mean recovery time was longer for subjects on Tyzeka than subjects on lamivudine.
- Among Tyzeka-treated subjects with Grade 1-4 CK elevations, 10% developed a musculoskeletal adverse event compared to 5% of lamivudine-treated subjects. A total of 2% (13/847) Tyzeka-treated subjects interrupted or discontinued trial drug due to CK elevation or musculoskeletal adverse events1.
- 1 Includes the Preferred Terms: back pain, chest wall pain, non-cardiac chest pain, chest discomfort, flank pain, muscle cramp, muscular weakness, musculoskeletal pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal stiffness, myalgia, myofascial pain syndrome, myopathy, myositis, neck pain, and pain in extremity.
- ALT Flares During Treatment
- The incidence of ALT flares, defined as ALT greater than 10 x ULN and greater than 2 x baseline, was similar in the two treatment arms (3%) in the first six months. After week 24, ALT flares were reported less frequently in the Tyzeka arm (2%) compared to the lamivudine arm (5%). Periodic monitoring of hepatic function is recommended during chronic hepatitis B treatment.
- Exacerbations of Hepatitis after Discontinuation of Treatment
- In the subset of subjects who discontinued treatment prematurely for reasons other than efficacy, or who elected not to continue Tyzeka in another clinical trial, 9/154 (6%) Tyzeka-treated and 10/180 (6%) lamivudine-treated subjects experienced an exacerbation of hepatitis (ALT elevation greater than 2 x baseline and greater than 10 x ULN) in the 4-month post-treatment period.
- Results at 208 Weeks
- After 104 weeks of blinded therapy in trials 007 GLOBE and NV-02B-015, 667 subjects received Tyzeka in an open-label extension trial, CLDT600A2303. Of those initially randomized to Tyzeka therapy, 78% of subjects (530/680) from trial 007 GLOBE and 82% (137/167) of subjects from trial NV-02B-015 enrolled into the extension trial and continued Tyzeka treatment for up to 208 weeks. The long-term Tyzeka safety population in trial CLDT600A2303 consisted of 655 subjects, including 518 subjects from trial 007 GLOBE and 137 subjects from trial NV-02B-015.
- The overall safety profile from the pooled analysis up to 104 and 208 weeks was similar. Grade 3/4 CK elevations occurred in 16% of subjects (104/655) treated with Tyzeka in trial CLDT600A2303. Most grade 3/4 CK elevations were asymptomatic (74% of subjects without any muscle related adverse reaction) and transient (98% of episodes lasted one or two visits (visit interval 2 - 12 weeks) and 87% of subjects had one or two episodes). Most grade 3/4 CK elevations (93%) resolved spontaneously or returned to baseline levels. Two cases of myopathy and two cases of myositis were reported in the 655 Tyzeka-treated subjects.
- Among the cohort of 655 subjects continuing Tyzeka for up to 208 weeks in trial CLDT600A2303, including the subgroup of patients (n=223) with mild renal impairment (eGFR 60-90 mL per min) at baseline, mean estimated GFR assessed by MDRD did not decline.
Postmarketing Experience
- The following adverse reactions have been reported during post approval use of Tyzeka. Because these reactions were reported voluntarily from a population of unknown size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal and Connective Tissue Disorders=
Nervous System Disorders
Metabolism and Nutrition Disorders
Drug Interactions
- Tyzeka is excreted mainly by passive diffusion so the potential for interactions between Tyzeka and other drugs eliminated by renal excretion is low. However, because Tyzeka is eliminated primarily by renal excretion, coadministration of Tyzeka with drugs that alter renal function may alter plasma concentrations of Tyzeka.
- A clinical trial investigating the combination of Tyzeka, 600 mg daily, with pegylated interferon alfa-2a, 180 micrograms once weekly by subcutaneous administration, indicates that this combination is associated with an increased risk of peripheral neuropathy occurrence and severity, in comparison to Tyzeka or pegylated interferon alfa-2a alone.
Use in Specific Populations
Pregnancy
- Pregnancy Category B
- Telbivudine is not teratogenic and has shown no adverse effects in developing embryos and fetuses in preclinical studies. Studies in pregnant rats and rabbits showed that telbivudine crosses the placenta. Developmental toxicity studies revealed no evidence of harm to the fetus in rats and rabbits at doses up to 1000 mg per kg per day, providing exposure levels 6- and 37-times higher, respectively, than those observed with the 600 mg per day dose in humans.
- There are no adequate and well-controlled trials of Tyzeka in pregnant women. Because animal reproductive toxicity studies are not always predictive of human response, Tyzeka should be used during pregnancy only if potential benefits outweigh the risks.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Telbivudine in women who are pregnant.
Labor and Delivery
- There are no trials in pregnant women and no data on the effect of Tyzeka on transmission of HBV from mother to infant. Therefore, appropriate interventions should be used to prevent neonatal acquisition of HBV infection.
Nursing Mothers
- Telbivudine is excreted in the milk of rats. It is not known whether Tyzeka is excreted in human milk. Mothers should be instructed not to breast-feed if they are receiving Tyzeka.
Pediatric Use
- Safety and effectiveness of Tyzeka in pediatric patients have not been established.
Geriatic Use
- Clinical trials of Tyzeka did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. In general, caution should be exercised when prescribing Tyzeka to elderly patients, considering the greater frequency of decreased renal function due to concomitant disease or other drug therapy. Renal function should be monitored in elderly patients, and dosage adjustments should be made accordingly.
Gender
There is no FDA guidance on the use of Telbivudine with respect to specific gender populations.
Race
- The safety and efficacy of Tyzeka have not been evaluated in Black/African American or Hispanic patients. It is not known if safety and efficacy can be extrapolated from studied populations.
Renal Impairment
- Tyzeka is eliminated primarily by renal excretion, therefore dose regimen adjustment is recommended in patients with creatinine clearance less than 50 mL per min, including patients with ESRD requiring hemodialysis.
Hepatic Impairment
There is no FDA guidance on the use of Telbivudine in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Telbivudine in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Telbivudine in patients who are immunocompromised.
Administration and Monitoring
Administration
- Oral
Monitoring
There is limited information regarding Monitoring of Telbivudine in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Telbivudine in the drug label.
Overdosage
Acute Overdose
Signs and Symptoms
- There is no information on intentional overdose of Tyzeka, but one subject experienced an unintentional and asymptomatic overdose. Healthy subjects who received Tyzeka doses up to 1800 mg per day for 4 days had no increase in or unexpected adverse events. A maximum tolerated dose for Tyzeka has not been determined. In the event of an overdose, Tyzeka should be discontinued, the patient must be monitored for evidence of toxicity, and appropriate general supportive treatment applied as necessary.
Management
- In case of overdosage, hemodialysis may be considered. Within 2 hours, following a single 200 mg dose of telbivudine, a 4-hour hemodialysis session removed approximately 23% of the telbivudine dose.
Chronic Overdose
There is limited information regarding Chronic Overdose of Telbivudine in the drug label.
Pharmacology

Mechanism of Action
- Telbivudine is a synthetic thymidine nucleoside analogue with activity against HBV DNA polymerase. It is phosphorylated by cellular kinases to the active triphosphate form, which has an intracellular half-life of 14 hours. Telbivudine 5'-triphosphate inhibits HBV DNA polymerase (reverse transcriptase) by competing with the natural substrate, thymidine 5'-triphosphate. Incorporation of telbivudine 5'-triphosphate into viral DNA causes DNA chain termination. Telbivudine is an inhibitor of both HBV first strand (EC50 value = 1.3 ± 1.6 micromolar) and second strand synthesis (EC50 value = 0.2 ± 0.2 micromolar). Telbivudine 5'-triphosphate at concentrations up to 100 micromolar did not inhibit human cellular DNA polymerases α, β, or γ. No appreciable mitochondrial toxicity was observed in HepG2 cells treated with telbivudine at concentrations up to 10 micromolar.
Structure
- Tyzeka is the trade name for telbivudine, a synthetic thymidine nucleoside analogue with activity against hepatitis B virus (HBV). The chemical name for telbivudine is 1-((2S,4R,5S)-4-hydroxy-5-hydroxymethyltetrahydrofuran-2-y1)-5-methyl-1H-pyrimidine-2,4-dione, or 1-(2-deoxy-β-L-ribofuranosyl)-5-methyluracil. Telbivudine is the unmodified β-L enantiomer of the naturally occurring nucleoside, thymidine. Its molecular formula is C10H14N2O5, which corresponds to a molecular weight of 242.23. Telbivudine has the following structural formula:
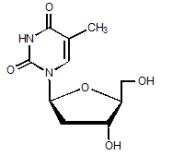
This image is provided by the National Library of Medicine.

- Telbivudine is a white to slightly yellowish powder. Telbivudine is sparingly soluble in water (greater than 20 mg per mL), and very slightly soluble in absolute ethanol (0.7 mg per mL) and n-octanol (0.1 mg per mL).
- Tyzeka film-coated tablets are available for oral administration in 600 mg strength. Tyzeka 600 mg film-coated tablets contain the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, povidone, and sodium starch glycolate. The tablet coating contains titanium dioxide, polyethylene glycol, talc, and hypromellose.
- Tyzeka oral solution is available for oral administration in 100 mg per 5 mL strength. Tyzeka oral solution contains the following inactive ingredients: citric acid anhydrous, benzoic acid, passion fruit flavor, sodium saccharin, sodium hydroxide, and purified water. A 600 mg dose (30 mL) of Tyzeka oral solution contains approximately 47 mg of sodium.
Pharmacodynamics
- In a randomized, partially single-blinded, placebo and active-controlled, four-period crossover trial, 53 healthy subjects were administered Tyzeka 600 mg, a supratherapeutic Tyzeka 1800 mg dose, placebo, and moxifloxacin 400 mg. After 7 days of dosing, Tyzeka did not prolong the QT interval. The maximum placebo-adjusted mean (upper 1-side 95% CI) change from baseline in QTcF on day 7 were 3.4 msec (5.9 msec) for the 600 mg and 4.4 msec (6.9 msec) for the 1800 mg dosing regimens.
Pharmacokinetics
- The single- and multiple-dose pharmacokinetics of Tyzeka were evaluated in healthy subjects and in patients with chronic hepatitis B. Tyzeka pharmacokinetics are similar between both populations.
- Absorption and Bioavailability
- Following oral administration of Tyzeka 600 mg once-daily in healthy subjects (n=12), steady state peak plasma concentration (Cmax) was 3.69 ± 1.25 microgram per mL (mean ± SD) which occurred between 1 and 4 hours (median 2 hours), AUC was 26.1 ± 7.2 microgram * hour per mL (mean ± SD), and trough plasma concentrations (Ctrough) were approximately 0.2-0.3 microgram per mL. Steady state was achieved after approximately 5 to 7 days of once-daily administration with ~1.5-fold accumulation, suggesting an effective half-life of ~15 hours.
- Effects of Food on Oral Absorption
- Tyzeka absorption and exposure were unaffected when a single 600 mg dose was administered with a high-fat (~55 g), high-calorie (~950 kcal) meal. Tyzeka may be taken with or without food.
- Distribution
- n vitro binding of telbivudine to human plasma proteins is low (3.3%). After oral dosing, the estimated apparent volume of distribution is in excess of total body water, suggesting that telbivudine is widely distributed into tissues. Telbivudine was equally partitioned between plasma and blood cells.
- Metabolism and Elimination
- No metabolites of telbivudine were detected following administration of [14C]-telbivudine in humans. Telbivudine is not a substrate, or inhibitor of the cytochrome P450 (CYP450) enzyme system.
- After reaching the peak concentration, plasma concentrations of Tyzeka declined in a biexponential manner with a terminal elimination half-life (T1/2) of 40-49 hours. Tyzeka is eliminated primarily by urinary excretion of unchanged drug. The renal clearance of Tyzeka approaches normal glomerular filtration rate suggesting that passive diffusion is the main mechanism of excretion. Approximately 42% of the dose is recovered in the urine over 7 days following a single 600 mg oral dose of Tyzeka. Because renal excretion is the predominant route of elimination, patients with moderate to severe renal dysfunction and those undergoing hemodialysis require a dose regimen adjustment.
- Special Populations
- Gender: There are no significant gender-related differences in Tyzeka pharmacokinetics.
- Race: There are no significant race-related differences in Tyzeka pharmacokinetics.
- Pediatrics and Geriatrics: Pharmacokinetic studies have not been conducted in children or elderly subjects.
- Renal Impairment: Single-dose pharmacokinetics of Tyzeka have been evaluated in subjects (without chronic hepatitis B) with various degrees of renal impairment (as assessed by creatinine clearance). Based on the results shown in Table 4, adjustment of the dose regimen for Tyzeka is recommended in patients with creatinine clearance of less than 50 mL per min.
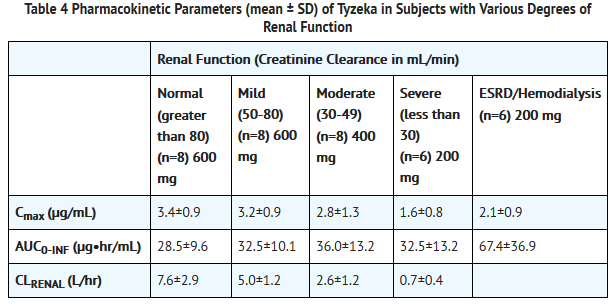
This image is provided by the National Library of Medicine.

- Renally Impaired Subjects on Hemodialysis: Hemodialysis (up to 4 hours) reduces systemic Tyzeka exposure by approximately 23%. Following dose regimen adjustment for creatinine clearance, no additional dose modification is necessary during routine hemodialysis. When administered on hemodialysis days, Tyzeka should be administered after hemodialysis.
- Hepatic Impairment: The pharmacokinetics of Tyzeka following a single 600 mg dose have been studied in subjects (without chronic hepatitis B) with various degrees of hepatic impairment. There were no changes in Tyzeka pharmacokinetics in hepatically impaired subjects compared to unimpaired subjects. Results of these studies indicate that no dosage adjustment is necessary for patients with hepatic impairment.
- Drug Interactions
- Drug-drug interaction studies show that lamivudine, adefovir dipivoxil, cyclosporine, pegylated interferon alfa-2a, and tenofovir disoproxil fumarate do not alter Tyzeka pharmacokinetics. In addition, Tyzeka does not alter the pharmacokinetics of lamivudine, adefovir dipivoxil, cyclosporine, or tenofovir disoproxil fumarate. No definitive conclusion could be drawn regarding the effects of Tyzeka on the pharmacokinetics of pegylated interferon alfa-2a due to the high inter-individual variability of pegylated interferon alfa-2a concentrations. At concentrations up to 12 times that in humans, telbivudine did not inhibit in vitro metabolism mediated by any of the following human hepatic microsomal cytochrome P450 (CYP) isoenzymes known to be involved in human medicinal product metabolism: 1A2, 2C9, 2C19, 2D26, 2E1, and 3A4. Based on the above results and the known elimination pathway of telbivudine, the potential for CYP450-mediated interactions involving telbivudine with other medicinal products is low.
Nonclinical Toxicology
- Telbivudine has shown no carcinogenic potential. Long term oral carcinogenicity studies with telbivudine were negative in mice and rats at exposures up to 14 times those observed in humans at the therapeutic dose of 600 mg per day.
- There was no evidence of genotoxicity based on in vitro or in vivo tests. Telbivudine was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains with or without metabolic activation. Telbivudine was not clastogenic in mammalian-cell gene mutation assays, including human lymphocyte cultures and an assay with Chinese hamster ovary cells with or without metabolic activation. Furthermore, telbivudine showed no effect in an in vivo micronucleus study in mice.
- Effects on fertility were studied in rats administered telbivudine as juveniles or adults. Juvenile rats were treated with telbivudine at doses of 0, 250, 1000, and 2000 mg per kg per day from post natal days 14 to 70. These rats were mated following a 5 week drug-free recovery period. Up to 50% reduction of fertility was associated with doses 1000 mg per kg per day and higher, which was equivalent to a systemic exposure approximately 7.5 times that achieved in humans at the therapeutic dose. The no observed adverse effect level (NOAEL) for effects on fertility or mating parameters was 250 mg per kg per day, which was equivalent to systemic exposure levels 2.5 to 2.8 times that achieved in humans at the therapeutic dose. In contrast, such reduction of fertility was absent in adult rats treated with telbivudine at doses up to 2000 mg per kg per day, equivalent to a systemic exposure approximately 14 times that achieved in humans at the therapeutic dose.
Clinical Studies
Clinical Experience in Nucleoside-Naïve Adults
- The safety and efficacy of long-term (104-week) Tyzeka treatment were evaluated in one active-controlled, clinical trial (NV-02B-007 GLOBE Trial) that included 1,367 subjects with chronic hepatitis B and a smaller supportive trial (NV-02B-015) that included 332 subjects. Subjects were 16 years of age or older, with chronic hepatitis B, evidence of HBV infection with viral replication (HBsAg-positive, HBeAg-positive or HBeAg-negative, HBV DNA detectable by a PCR assay), and elevated ALT levels greater than or equal to 1.3 x ULN, no evidence of hepatic decompensation, and chronic inflammation on liver biopsy compatible with chronic viral hepatitis.
- NV-02B-007 GLOBE Trial
- The Week 52 and Week 104 results of the 007 GLOBE trial are summarized below.
- The 007 GLOBE trial was a Phase III, randomized, double-blind, multinational trial of Tyzeka 600 mg once daily compared to lamivudine 100 mg once daily for a treatment period of 104 weeks in 1,367 (n= 680 Tyzeka; n=687 lamivudine) nucleoside-naïve chronic hepatitis B HBeAg-positive and HBeAg-negative subjects. The primary data analysis was conducted after all subjects had reached Week 52.
- HBeAg-positive Subjects: (n= 458 Tyzeka; n= 463 lamivudine) The mean age of subjects was 32 years, 74% were male, 82% were Asian, 12% were Caucasian, and 6% had previously received alfa-interferon therapy. At baseline, subjects had a mean Knodell Necroinflammatory Score greater than or equal to 7; mean serum HBV DNA as measured by Roche COBAS Amplicor® PCR assay was 9.52 log10 copies per mL; and mean serum ALT was 153 IU per L. Pre- and post-liver biopsy samples were adequate for 86% of subjects.
- HBeAg-negative Subjects: (n=222 Tyzeka; n= 224 lamivudine)The mean age of subjects was 43 years, 77% were male, 65% were Asian, 23% were Caucasian, and 11% had previously received alfa-interferon therapy. At baseline, subjects had a mean Knodell Necroinflammatory Score greater than or equal to 7; mean serum HBV DNA as measured by Roche COBAS Amplicor® PCR assay was 7.54 log10 copies per mL; and mean serum ALT was 140 IU per L. Pre- and post-liver biopsy samples were adequate for 92% of subjects.
- Clinical Results
- Clinical and virologic efficacy endpoints were evaluated separately in the HBeAg-positive and HBeAg-negative subject populations.
- The primary endpoint of Therapeutic Response at Week 52 was a composite endpoint requiring suppression of HBV DNA to less than 5 log10 copies per mL in conjunction with either loss of serum HBeAg or ALT normalization. Key secondary endpoints included histologic response, ALT normalization, and measures of virologic response.
- At Week 52, in HBeAg-positive subjects, 75% of Tyzeka subjects and 67% of lamivudine subjects had a Therapeutic Response; in HBeAg-negative subjects, 75% of Tyzeka subjects and 77% of lamivudine subjects had a Therapeutic Response.
- Analysis of the histological response at Week 52 is shown in Table 6.
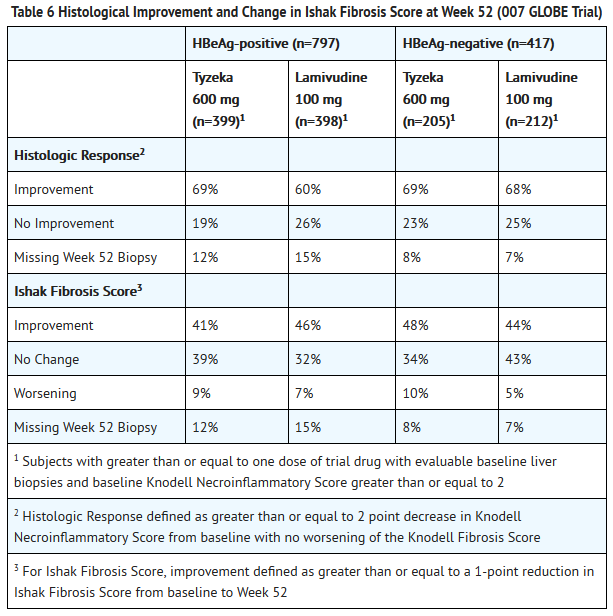
This image is provided by the National Library of Medicine.

- Subjects were eligible to continue blinded treatment to Week 104. In the ITT population, 624/680 (92%) Tyzeka recipients and 599/687 (87%) lamivudine recipients completed trial treatment to Week 104. At Week 104, in HBeAg-positive subjects, 63% of Tyzeka subjects and 48% of lamivudine subjects had a Therapeutic Response, while in HBeAg-negative subjects 78% of Tyzeka subjects and 66% of lamivudine subjects had a Therapeutic Response.
- Selected virologic, biochemical, and serologic outcome measures at Weeks 52 and 104 are shown in Table 7.

This image is provided by the National Library of Medicine.

- NV-02B-015 Trial
- The efficacy results of the 007 GLOBE trial were supported by results of trial NV-02B-015. This was a Phase III, randomized, double-blind, trial of Tyzeka 600 mg once daily compared to lamivudine 100 mg once daily for a treatment period of 104 weeks in 332 (n=167 Tyzeka; n=165 lamivudine) nucleoside-naïve chronic hepatitis B HBeAg-positive and HBeAg-negative Chinese subjects. The primary efficacy endpoint was serum HBV DNA reduction from baseline. In this trial, the composite endpoint Therapeutic Response was a key secondary endpoint. Histological response was not assessed as an outcome measure in this trial.
- Clinical Results
- Among HBeAg-positive subjects (n=147 Tyzeka; n=143 lamivudine) results for key endpoints at Week 104 included Therapeutic Response (66% vs. 41%), mean HBV DNA reduction (-5.47 vs. -3.97 log10 copies per mL), HBV DNA PCR negativity (58% vs. 34%), ALT normalization (73% vs. 59%), HBeAg loss (40% vs. 28%) and HBeAg seroconversion (29% vs. 20%), for Tyzeka and lamivudine, respectively. Because the number of HBeAg-negative subjects in this trial was small (n=42), definitive conclusions could not be drawn regarding efficacy outcomes in this subpopulation.
How Supplied
- Tablets
- Tyzeka 600 mg tablets are white to slightly yellowish film-coated, ovaloid-shaped tablets, imprinted with “LDT” on one side.
- tle of 30 tablets (NDC 0078-0538-15) with child-resistant closure.
- Store Tyzeka tablets in original container at 25ºC (77ºF), excursions permitted to 15-30ºC (59-86ºF).
- Oral Solution
- Tyzeka (telbivudine) oral solution is a clear, colorless to pale yellow, passion fruit flavored liquid. Tyzeka oral solution contains 100 mg of telbivudine per 5 milliliters.
- Bottle containing 300 mL oral solution (NDC 0078-0539-85) with child-resistant closure and embossed dosing cup. The dosing cup is intended for measurement of Tyzeka oral solution only.
- Store Tyzeka oral solution in original container at 25ºC (77ºF), excursions permitted to 15-30ºC (59-86ºF). Use within two months after opening the bottle. Do not freeze.
- For all medical inquiries call: 1-877-8-Tyzeka (1-877-889-9352).
- Keep this and all drugs out of the reach of children.
Storage
There is limited information regarding Telbivudine Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Telbivudine |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Telbivudine |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- Patients should remain under the care of a physician while taking Tyzeka. They should discuss any new symptoms or concurrent medications with their physician.
- Patients should be advised to report promptly unexplained muscle weakness, tenderness, or pain.
- Patients should be advised to report promptly any numbness, tingling, and/or burning sensations in the arms and/or legs, with or without difficulty walking.
- Patients should be advised that Tyzeka is not a cure for hepatitis B, that the long-term treatment benefits of Tyzeka are unknown at this time. In particular, the relationship of initial treatment response to outcomes such as hepatocellular carcinoma and decompensated cirrhosis is unknown.
- Patients should be informed that deterioration of liver disease may occur in some cases if treatment is discontinued, and that they should discuss any change in regimen with their physician.
- Patients should be advised that treatment with Tyzeka has not been shown to reduce the risk of transmission of HBV to others through sexual contact or blood contamination. HBV prevention strategies should be discussed with patients, including safe sexual practices, and avoidance of needle sharing or sharing any personal items which may contain residual blood or body fluids, such as razor blades or toothbrushes. Additionally, a vaccine is available for prevention of hepatitis B infection in susceptible individuals.
- Patients on a low sodium diet should be advised that Tyzeka oral solution contains approximately 47 mg of sodium per 600 mg dose (30 mL).
- Patients should be advised to dispose of unused or expired Tyzeka by using a community pharmaceutical take-back disposal program, or by placing unused Tyzeka in a closed container, such as a sealed bag, into household trash. All identifying information should be removed from the original Tyzeka container prior to disposal.

This image is provided by the National Library of Medicine.

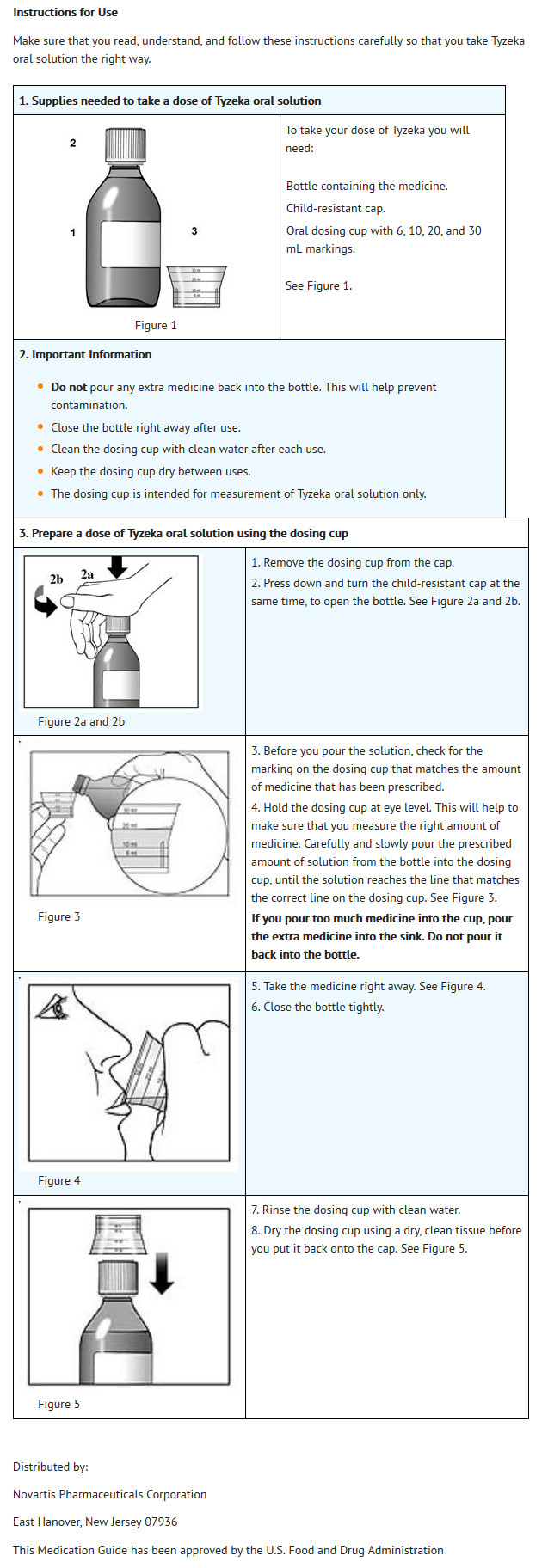
This image is provided by the National Library of Medicine.

Precautions with Alcohol
- Alcohol-Telbivudine interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- TYZEKA®[1]
Look-Alike Drug Names
There is limited information regarding Telbivudine Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Telbivudine
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Telbivudine |Label Name=Telbivudine10.png
}}
{{#subobject:
|Label Page=Telbivudine |Label Name=Telbivudine11.png
}}
{{#subobject:
|Label Page=Telbivudine |Label Name=Telbivudine12.png
}}