Glycogen storage disease
|
Glycogen storage disease |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Anmol Pitliya, M.B.B.S. M.D.[2], Cafer Zorkun, M.D., Ph.D. [3]
Synonyms and keywords:Glycogenosis; dextrinosis
Overview
Glycogen storage diseases are several inborn errors of metabolism that result from enzyme defects that affect the processing of glycogen synthesis or breakdown within muscles, liver, and other cell types. A total of fourteen glycogen storage diseases have been described which differ from each other on the basis of genotypic and phenotypic heterogenity. Most of the glycogen storage diseases follow an autosomal recessive mode of inheritence. In 1929, Von Gierke was the first to describe glycogen storage disease in a 8 year old girl.[1] Glucose-6-phosphatase deficiency found in glycogen storage disease type I is identified as first specific enzymopathy in a hereditary disorder.[2] Majority of glycogen storage diseases are due to deficiency of specific enzymes involved in metabolism of glycogen either in liver or muscle or both. These deficiencies commonly result in excess of glycogen which deposits in several tissues in the body. There are a wide variety of clinical manifestations of glycogen storage diseases. However, common clinical manifestations of various glycogen storage diseases include hypoglycemia, hypotonia, muscle weakness, hepatomegaly, cardiomegaly, elevated creatine kinase, hyperlipidemia, myoglobinuria, and elevated liver aminotransferases.
Pathophysiology
Metabolic Pathway

Gross Pathological Findings
Images shown below are courtesy of Professor Peter Anderson DVM PhD and published with permission. © PEIR, University of Alabama at Birmingham, Department of Pathology
-
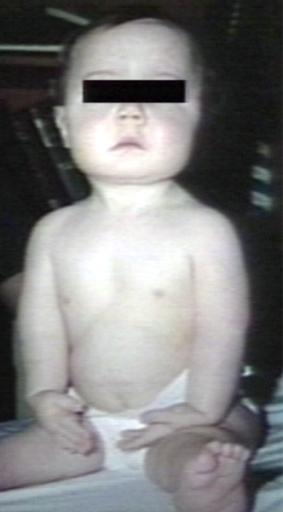
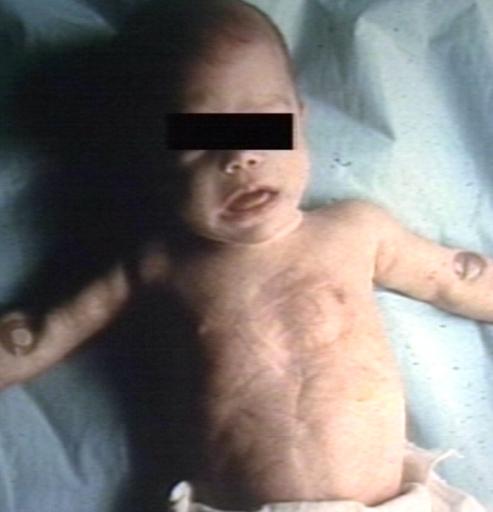 Pompe's Disease, Glycogen Storage Disease Type II. Child in crib
Pompe's Disease, Glycogen Storage Disease Type II. Child in crib -
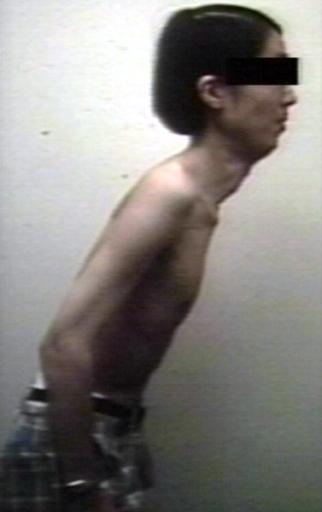 Pompe's Disease, Glycogen Storage Disease Type II
Pompe's Disease, Glycogen Storage Disease Type II -
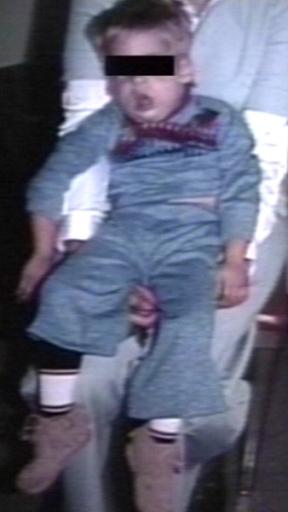 Pompe's Disease, Glycogen Storage Disease Type II
Pompe's Disease, Glycogen Storage Disease Type II



-
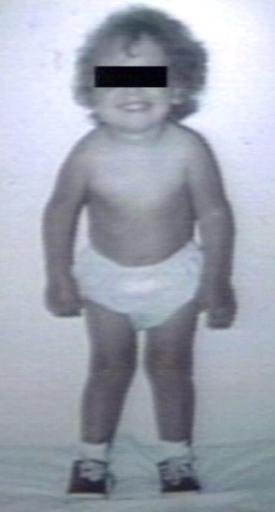 Pompe's Disease, Glycogen Storage Disease Type II, 9 years old patient
Pompe's Disease, Glycogen Storage Disease Type II, 9 years old patient -
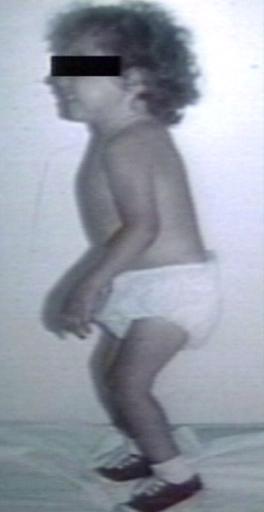 Pompe's Disease, Glycogen Storage Disease Type II, 9 years old patient
Pompe's Disease, Glycogen Storage Disease Type II, 9 years old patient


-
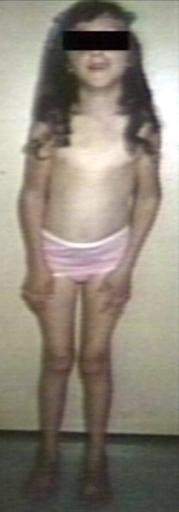 Pompe's Disease, Glycogen Storage Disease Type II
Pompe's Disease, Glycogen Storage Disease Type II -
 Pompe's Disease, Glycogen Storage Disease Type II
Pompe's Disease, Glycogen Storage Disease Type II


Microscopic Pathological Findings
Images shown below are courtesy of Professor Peter Anderson DVM PhD and published with permission. © PEIR, University of Alabama at Birmingham, Department of Pathology
-
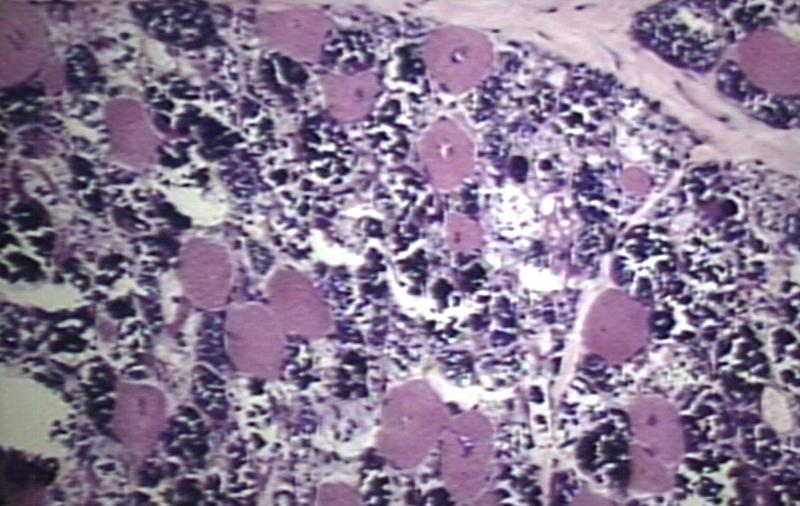 Muscle: Glycogen Storage Disease
Muscle: Glycogen Storage Disease -
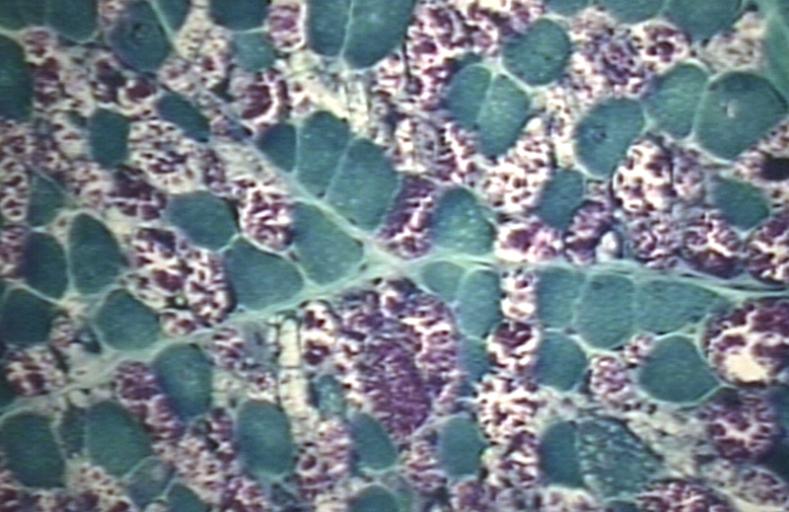 Muscle: Glycogen Storage Disease
Muscle: Glycogen Storage Disease -
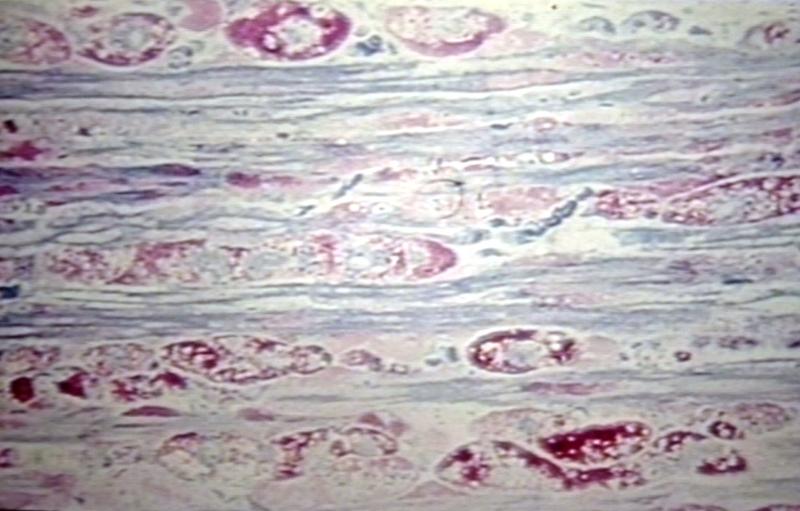 Nerve: Glycogen Storage Disease Macrophages; Longitudinal Section of Peripheral Nerve
Nerve: Glycogen Storage Disease Macrophages; Longitudinal Section of Peripheral Nerve



Differentiating various Glycogen Storage Diseases
| Differentiating Glycogen Storage Diseases | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glycogen storage disease | Enzyme deficiency | Genetics | History and symptoms | Physical examination | Laboratory findings | Imaging | Other features | |||||||
| Gene mutation | Inheritance | Chromosome | Hypoglycemia | Muscle weakness | Hypotonia | Hepatomegaly | Elevated CK | Cardiomegaly | ||||||
| Glycogen storage disease type I[3][4][5][6][7][8][9] | Von Gierke's disease | GSD type Ia | Glucose-6-phosphatase | G6PC gene mutation | Autosomal recessive | 17q21 | + | + | + | + | - | - | ||
| GSD type Ib | Microsomal glucose-6-phosphate transporter | SLC37A4 gene mutation | Autosomal recessive | 11q23 | ||||||||||
| Glycogen storage disease type II[10][11][12][13][14][15][16][17][18] | Pompe disease | Infantile onset | Acid alpha-glucosidase | GAA gene | Autosomal recessive | 17q25 | - | + | + | + | + | + |
| |
| Late onset | Autosomal recessive | - | + | + | + | + | +/- | |||||||
| Glycogen storage disease type III[19][20][21][22][23][24] | Cori disease | GSD type IIIa | Debranching enzyme (deficiency in muscle and liver) | AGL gene mutation | Autosomal recessive | 1p21 | + | + | + | + | + | + |
| |
| GSD type IIIb | Debranching enzyme (deficiency in liver only) | Autosomal recessive | ||||||||||||
| Glycogen storage disease type IV[25][26][27][28][29] | Andersen's disease | Branching enzyme | GBE1 gene mutation | Autosomal recessive | 3p12 | +/- | + | + | + | + | + | - | ||
| Glycogen storage disease type V[30][31][32][33][34][35][36] | McArdle disease | Muscle glycogen phosphorylase | PYGM gene mutation | Autosomal recessive | 11q13 | - | + | - | - | + | - |
| ||
| Glycogen storage disease type VI[37][38][39][40][41] | Hers' disease | Autosomal | Liver glycogen phosphorylase | PYGL gene mutation | Autosomal recessive | 14q22 | +/- | + | +/- | + | - | - |
| |
| X-linked | PYGL gene mutation | X-linked recessive | X | |||||||||||
| Glycogen storage disease type VII[42][43][44][45][46][47] | Tarui's disease | Muscle phosphofructokinase | PFKM gene mutation | Autosomal recessive | 12q13 | + | + | - | - | + | + | |||
| Glycogen storage disease type IX[48][38][49] | GSD type IXa[50][51][52][53][54] | Phosphorylase b kinase (deficiency in liver only) | PHKA2 gene mutation | X-linked recessive | Xp22 | + | - | - | + | - | - |
| ||
| GSD type IXb[55][56][57] | Phosphorylase b kinase (deficiency in liver and muscle) | PHKB gene mutation | Autosomal recessive | 16q12 | + | - | - | + | - | - |
| |||
| Glycogen storage disease type X[58][59][60][61] | Phosphoglycerate mutase | PGAM2 gene mutation | Autosomal recessive | 7p13 | - | - | - | - | + | - |
| |||
| Glycogen storage disease type XI[62][63][64][65] | Lactate dehydrogenase A deficiency | Lactate dehydrogenase A | LDHA gene mutation | Autosomal recessive | 11p15 | - | - | - | - | + | - |
| ||
| Glycogen storage disease type XII[66][67][68][69] | Aldolase A deficiency | Aldolase A | ALDOA gene mutation | Autosomal recessive | 16p11 | - | + | - | + | - | - | |||
| Glycogen storage disease type XIII[70] | Beta-enolase | ENO3 gene mutation | Autosomal recessive | 17p13 | - | + | - | - | + | - | - | |||
| Glycogen storage disease type XIV[71][72] | Phosphoglucomutase type 2 | PGM1 gene mutation | Autosomal recessive | 1p31 | +/- | + | - | - | + | - |
| |||
| Glycogen storage disease type 0[73][74][75][76] | Lewis' disease | Hepatic glycogen synthase | GYS2 gene mutation (liver) | Autosomal recessive | 12p12 | + | - | - | - | - | - |
| ||
Heart & Liver in Glycogen Storage Disease
{{#ev:youtube|inSkXkNK_dE}}
References
- ↑ Gierke E, Von (1929). "Hepato-nephro-megalia-glycogenica". Beitr Pathol Anat. 82: 497–513.
- ↑ Ozen H (2007). "Glycogen storage diseases: new perspectives". World J Gastroenterol. 13 (18): 2541–53. PMC 4146814. PMID 17552001.
- ↑ Mansfield BC (1999). "Molecular Genetics of Type 1 Glycogen Storage Diseases". Trends Endocrinol Metab. 10 (3): 104–113. PMID 10322403.
- ↑ Ozen H (2007). "Glycogen storage diseases: new perspectives". World J Gastroenterol. 13 (18): 2541–53. PMC 4146814. PMID 17552001.
- ↑ Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A; et al. (2011). "Glucose-6-phosphatase deficiency". Orphanet J Rare Dis. 6: 27. doi:10.1186/1750-1172-6-27. PMC 3118311. PMID 21599942.
- ↑ Kishnani, Priya S.; Austin, Stephanie L.; Abdenur, Jose E.; Arn, Pamela; Bali, Deeksha S.; Boney, Anne; Chung, Wendy K.; Dagli, Aditi I.; Dale, David; Koeberl, Dwight; Somers, Michael J.; Burns Wechsler, Stephanie; Weinstein, David A.; Wolfsdorf, Joseph I.; Watson, Michael S. (2014). "Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics". Genetics in Medicine. doi:10.1038/gim.2014.128. ISSN 1098-3600.
- ↑ Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP (2002). "Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I)". Eur. J. Pediatr. 161 Suppl 1: S20–34. doi:10.1007/s00431-002-0999-4. PMID 12373567.
- ↑ Bali DS, Chen YT, Austin S, et al. Glycogen Storage Disease Type I. 2006 Apr 19 [Updated 2016 Aug 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1312/
- ↑ Griggs, Robert (2014). Evaluation and treatment of myopathies. Oxford: Oxford University Press. ISBN 9780199873944.
- ↑ Leslie N, Bailey L. Pompe Disease. 2007 Aug 31 [Updated 2017 May 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1261/
- ↑ Di Rocco M, Buzzi D, Tarò M (2007). "Glycogen storage disease type II: clinical overview". Acta Myol. 26 (1): 42–4. PMC 2949314. PMID 17915568.
- ↑ Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D; et al. (2006). "A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease". J Pediatr. 148 (5): 671–676. doi:10.1016/j.jpeds.2005.11.033. PMID 16737883.
- ↑ van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT; et al. (2003). "The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature". Pediatrics. 112 (2): 332–40. PMID 12897283.
- ↑ Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F (2000). "Identification of two subtypes of infantile acid maltase deficiency". J Pediatr. 137 (2): 283–5. doi:10.1067/mpd.2000.107112. PMID 10931430.
- ↑ Martiniuk F, Mehler M, Tzall S, Meredith G, Hirschhorn R (1990). "Sequence of the cDNA and 5'-flanking region for human acid alpha-glucosidase, detection of an intron in the 5' untranslated leader sequence, definition of 18-bp polymorphisms, and differences with previous cDNA and amino acid sequences". DNA Cell Biol. 9 (2): 85–94. doi:10.1089/dna.1990.9.85. PMID 2111708.
- ↑ Hoefsloot LH, Hoogeveen-Westerveld M, Kroos MA, van Beeumen J, Reuser AJ, Oostra BA (1988). "Primary structure and processing of lysosomal alpha-glucosidase; homology with the intestinal sucrase-isomaltase complex". EMBO J. 7 (6): 1697–704. PMC 457155. PMID 3049072.
- ↑ Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA (1990). "Characterization of the human lysosomal alpha-glucosidase gene". Biochem J. 272 (2): 493–7. PMC 1149727. PMID 2268276.
- ↑ Kuo WL, Hirschhorn R, Huie ML, Hirschhorn K (1996). "Localization and ordering of acid alpha-glucosidase (GAA) and thymidine kinase (TK1) by fluorescence in situ hybridization". Hum Genet. 97 (3): 404–6. PMID 8786092.
- ↑ Shen J, Bao Y, Liu HM, Lee P, Leonard JV, Chen YT (1996). "Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle". J Clin Invest. 98 (2): 352–7. doi:10.1172/JCI118799. PMC 507437. PMID 8755644.
- ↑ Ding JH, de Barsy T, Brown BI, Coleman RA, Chen YT (1990). "Immunoblot analyses of glycogen debranching enzyme in different subtypes of glycogen storage disease type III". J Pediatr. 116 (1): 95–100. PMID 2295969.
- ↑ Aoyama Y, Ozer I, Demirkol M, Ebara T, Murase T, Podskarbi T; et al. (2009). "Molecular features of 23 patients with glycogen storage disease type III in Turkey: a novel mutation p.R1147G associated with isolated glucosidase deficiency, along with 9 AGL mutations". J Hum Genet. 54 (11): 681–6. doi:10.1038/jhg.2009.100. PMID 19834502.
- ↑ Kishnani, Priya S; Austin, Stephanie L; Arn, Pamela; Bali, Deeksha S; Boney, Anne; Case, Laura E; Chung, Wendy K; Desai, Dev M; El-Gharbawy, Areeg; Haller, Ronald; Smit, G Peter A; Smith, Alastair D; Hobson-Webb, Lisa D; Wechsler, Stephanie Burns; Weinstein, David A; Watson, Michael S (2010). "Glycogen Storage Disease Type III diagnosis and management guidelines". Genetics in Medicine. 12 (7): 446–463. doi:10.1097/GIM.0b013e3181e655b6. ISSN 1098-3600.
- ↑ Dagli A, Sentner CP, Weinstein DA. Glycogen Storage Disease Type III. 2010 Mar 9 [Updated 2016 Dec 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26372/
- ↑ Wolfsdorf JI, Weinstein DA (2003). "Glycogen storage diseases". Rev Endocr Metab Disord. 4 (1): 95–102. PMID 12618563.
- ↑ Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffrè B, Donati MA; et al. (2004). "Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV)". Neurology. 63 (6): 1053–8. PMID 15452297.
- ↑ Bruno C, Cassandrini D, Assereto S, Akman HO, Minetti C, Di Mauro S (2007). "Neuromuscular forms of glycogen branching enzyme deficiency". Acta Myol. 26 (1): 75–8. PMC 2949312. PMID 17915577.
- ↑ Brown BI, Brown DH (1966). "Lack of an alpha-1,4-glucan: alpha-1,4-glucan 6-glycosyl transferase in a case of type IV glycogenosis". Proc Natl Acad Sci U S A. 56 (2): 725–9. PMC 224432. PMID 5229990.
- ↑ McConkie-Rosell A, Wilson C, Piccoli DA, Boyle J, DeClue T, Kishnani P; et al. (1996). "Clinical and laboratory findings in four patients with the non-progressive hepatic form of type IV glycogen storage disease". J Inherit Metab Dis. 19 (1): 51–8. PMID 8830177.
- ↑ Magoulas PL, El-Hattab AW. Glycogen Storage Disease Type IV. 2013 Jan 3. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK115333/
- ↑ McARDLE B (1951). "Myopathy due to a defect in muscle glycogen breakdown". Clin Sci. 10 (1): 13–35. PMID 24540673.
- ↑ SCHMID R, MAHLER R (1959). "Chronic progressive myopathy with myoglobinuria: demonstration of a glycogenolytic defect in the muscle". J Clin Invest. 38: 2044–58. doi:10.1172/JCI103983. PMC 441792. PMID 14442994.
- ↑ Mommaerts WF, Illingworth B, Pearson CM, Guillory RJ, Seraydarian K (1959). "A FUNCTIONAL DISORDER OF MUSCLE ASSOCIATED WITH THE ABSENCE OF PHOSPHORYLASE". Proc Natl Acad Sci U S A. 45 (6): 791–7. PMC 222638. PMID 16590445.
- ↑ PEARSON CM, RIMER DG, MOMMAERTS WF (1961). "A metabolic myopathy due to absence of muscle phosphorylase". Am J Med. 30: 502–17. PMID 13733779.
- ↑ Grünfeld JP, Ganeval D, Chanard J, Fardeau M, Dreyfus JC (1972). "Acute renal failure in McArdle's disease. Report of two cases". N Engl J Med. 286 (23): 1237–41. doi:10.1056/NEJM197206082862304. PMID 4502558.
- ↑ Schmidt B, Servidei S, Gabbai AA, Silva AC, de Sousa Bulle de Oliveira A, DiMauro S (1987). "McArdle's disease in two generations: autosomal recessive transmission with manifesting heterozygote". Neurology. 37 (9): 1558–61. PMID 3476861.
- ↑ Martín MA, Lucía A, Arenas J, et al. Glycogen Storage Disease Type V. 2006 Apr 19 [Updated 2014 Jun 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1344/
- ↑ Wallis PG, Sidbury JB, Harris RC (1966). "Hepatic phosphorylase defect. Studies on peripheral blood". Am J Dis Child. 111 (3): 278–82. PMID 5904467.
- ↑ 38.0 38.1 Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J; et al. (2014). "The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada". Mol Genet Metab. 113 (3): 171–6. doi:10.1016/j.ymgme.2014.09.005. PMID 25266922.
- ↑ Burwinkel B, Bakker HD, Herschkovitz E, Moses SW, Shin YS, Kilimann MW (1998). "Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI". Am J Hum Genet. 62 (4): 785–91. PMC 1377030. PMID 9529348.
- ↑ Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG (1998). "Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI". Hum Mol Genet. 7 (5): 865–70. PMID 9536091.
- ↑ Dagli AI, Weinstein DA. Glycogen Storage Disease Type VI. 2009 Apr 23 [Updated 2011 May 17]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK5941/
- ↑ Raben N, Sherman JB (1995). "Mutations in muscle phosphofructokinase gene". Hum Mutat. 6 (1): 1–6. doi:10.1002/humu.1380060102. PMID 7550225.
- ↑ TARUI S, OKUNO G, IKURA Y, TANAKA T, SUDA M, NISHIKAWA M (1965). "PHOSPHOFRUCTOKINASE DEFICIENCY IN SKELETAL MUSCLE. A NEW TYPE OF GLYCOGENOSIS". Biochem Biophys Res Commun. 19: 517–23. PMID 14339001.
- ↑ Layzer RB, Rowland LP, Ranney HM (1967). "Muscle phosphofructokinase deficiency". Arch Neurol. 17 (5): 512–23. PMID 4228297.
- ↑ Satoyoshi E, Kowa H (1967). "A myopathy due to glycolytic abnormality". Arch Neurol. 17 (3): 248–56. PMID 4228753.
- ↑ Waterbury L, Frenkel EP (1972). "Hereditary nonspherocytic hemolysis with erythrocyte phosphofructokinase deficiency". Blood. 39 (3): 415–25. PMID 4258222.
- ↑ Vora S, Corash L, Engel WK, Durham S, Seaman C, Piomelli S (1980). "The molecular mechanism of the inherited phosphofructokinase deficiency associated with hemolysis and myopathy". Blood. 55 (4): 629–35. PMID 6444532.
- ↑ Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P; et al. (2007). "Glycogen storage disease type IX: High variability in clinical phenotype". Mol Genet Metab. 92 (1–2): 88–99. doi:10.1016/j.ymgme.2007.06.007. PMID 17689125.
- ↑ Goldstein J, Austin S, Kishnani P, et al. Phosphorylase Kinase Deficiency. 2011 May 31. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK55061/
- ↑ Keating JP, Brown BI, White NH, DiMauro S (1985). "X-linked glycogen storage disease. A cause of hypotonia, hyperuricemia, and growth retardation". Am J Dis Child. 139 (6): 609–13. PMID 3859203.
- ↑ Hendrickx J, Coucke P, Hors-Cayla MC, Smit GP, Shin YS, Deutsch J; et al. (1994). "Localization of a new type of X-linked liver glycogenosis to the chromosomal region Xp22 containing the liver alpha-subunit of phosphorylase kinase (PHKA2)". Genomics. 21 (3): 620–5. PMID 7959740.
- ↑ Schimke RN, Zakheim RM, Corder RC, Hug G (1973). "Glycogen storage disease type IX: benign glycogenosis of liver and hepatic phosphorylase kinase deficiency". J Pediatr. 83 (6): 1031–4. PMID 4518931.
- ↑ Willems PJ, Gerver WJ, Berger R, Fernandes J (1990). "The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients". Eur J Pediatr. 149 (4): 268–71. PMID 2303074.
- ↑ Hendrickx J, Bosshard NU, Willems P, Gitzelmann R (1998). "Clinical, biochemical and molecular findings in a patient with X-linked liver glycogenosis followed for 40 years". Eur J Pediatr. 157 (11): 919–23. PMID 9835437.
- ↑ Bashan N, Iancu TC, Lerner A, Fraser D, Potashnik R, Moses SW (1981). "Glycogenosis due to liver and muscle phosphorylase kinase deficiency". Pediatr Res. 15 (4 Pt 1): 299–303. doi:10.1203/00006450-198104000-00002. PMID 6938920.
- ↑ Gray RG, Kumar D, Whitfield AE (1983). "Glycogen phosphorylase b kinase deficiency in three siblings". J Inherit Metab Dis. 6 (3): 107. PMID 6422139.
- ↑ Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS; et al. (1997). "Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB)". Hum Mol Genet. 6 (7): 1109–15. PMID 9215682.
- ↑ Hadjigeorgiou GM, Kawashima N, Bruno C, Andreu AL, Sue CM, Rigden DJ; et al. (1999). "Manifesting heterozygotes in a Japanese family with a novel mutation in the muscle-specific phosphoglycerate mutase (PGAM-M) gene". Neuromuscul Disord. 9 (6–7): 399–402. PMID 10545043.
- ↑ Tsujino S, Shanske S, Sakoda S, Fenichel G, DiMauro S (1993). "The molecular genetic basis of muscle phosphoglycerate mutase (PGAM) deficiency". Am J Hum Genet. 52 (3): 472–7. PMC 1682163. PMID 8447317.
- ↑ Kissel JT, Beam W, Bresolin N, Gibbons G, DiMauro S, Mendell JR (1985). "Physiologic assessment of phosphoglycerate mutase deficiency: incremental exercise test". Neurology. 35 (6): 828–33. PMID 2987758.
- ↑ DiMauro S, Miranda AF, Khan S, Gitlin K, Friedman R (1981). "Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy". Science. 212 (4500): 1277–9. PMID 6262916.
- ↑ Yoshikuni K, Tagami H, Yamada M, Sudo K, Kanno T (1986). "Erythematosquamous skin lesions in hereditary lactate dehydrogenase M-subunit deficiency". Arch Dermatol. 122 (12): 1420–4. PMID 3789777.
- ↑ Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K (1988). "Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy". Clin Chim Acta. 173 (1): 89–98. PMID 3383424.
- ↑ Maekawa M, Sudo K, Kanno T (1986). "Immunochemical studies on lactate dehydrogenase A subunit deficiencies". Am J Hum Genet. 39 (2): 232–8. PMC 1683931. PMID 3092644.
- ↑ Takayasu S, Fujiwara S, Waki T (1991). "Hereditary lactate dehydrogenase M-subunit deficiency: lactate dehydrogenase activity in skin lesions and in hair follicles". J Am Acad Dermatol. 24 (2 Pt 2): 339–42. PMID 1999544.
- ↑ Kishi H, Mukai T, Hirono A, Fujii H, Miwa S, Hori K (1987). "Human aldolase A deficiency associated with a hemolytic anemia: thermolabile aldolase due to a single base mutation". Proc Natl Acad Sci U S A. 84 (23): 8623–7. PMC 299598. PMID 2825199.
- ↑ Beutler E, Scott S, Bishop A, Margolis N, Matsumoto F, Kuhl W (1973). "Red cell aldolase deficiency and hemolytic anemia: a new syndrome". Trans Assoc Am Physicians. 86: 154–66. PMID 4788792.
- ↑ Kreuder J, Borkhardt A, Repp R, Pekrun A, Göttsche B, Gottschalk U; et al. (1996). "Brief report: inherited metabolic myopathy and hemolysis due to a mutation in aldolase A." N Engl J Med. 334 (17): 1100–4. doi:10.1056/NEJM199604253341705. PMID 8598869.
- ↑ Hurst JA, Baraitser M, Winter RM (1987). "A syndrome of mental retardation, short stature, hemolytic anemia, delayed puberty, and abnormal facial appearance: similarities to a report of aldolase A deficiency". Am J Med Genet. 28 (4): 965–70. doi:10.1002/ajmg.1320280423. PMID 3688035.
- ↑ Comi GP, Fortunato F, Lucchiari S, Bordoni A, Prelle A, Jann S; et al. (2001). "Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis". Ann Neurol. 50 (2): 202–7. PMID 11506403.
- ↑ Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S; et al. (2014). "Multiple phenotypes in phosphoglucomutase 1 deficiency". N Engl J Med. 370 (6): 533–42. doi:10.1056/NEJMoa1206605. PMC 4373661. PMID 24499211.
- ↑ Stojkovic T, Vissing J, Petit F, Piraud M, Orngreen MC, Andersen G; et al. (2009). "Muscle glycogenosis due to phosphoglucomutase 1 deficiency". N Engl J Med. 361 (4): 425–7. doi:10.1056/NEJMc0901158. PMID 19625727.
- ↑ Orho M, Bosshard NU, Buist NR, Gitzelmann R, Aynsley-Green A, Blümel P; et al. (1998). "Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0". J Clin Invest. 102 (3): 507–15. doi:10.1172/JCI2890. PMC 508911. PMID 9691087.
- ↑ Laberge AM, Mitchell GA, van de Werve G, Lambert M (2003). "Long-term follow-up of a new case of liver glycogen synthase deficiency". Am J Med Genet A. 120A (1): 19–22. doi:10.1002/ajmg.a.20110. PMID 12794686.
- ↑ Gitzelmann R, Spycher MA, Feil G, Müller J, Seilnacht B, Stahl M; et al. (1996). "Liver glycogen synthase deficiency: a rarely diagnosed entity". Eur J Pediatr. 155 (7): 561–7. PMID 8831078.
- ↑ Rutledge SL, Atchison J, Bosshard NU, Steinmann B (2001). "Case report: liver glycogen synthase deficiency--a cause of ketotic hypoglycemia". Pediatrics. 108 (2): 495–7. PMID 11483824.