Enfuvirtide
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vignesh Ponnusamy, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Enfuvirtide is a HIV-1 fusion inhibitor that is FDA approved for the treatment of HIV-1 infection in treatment-experienced patients with HIV-1 replication despite ongoing antiretroviral therapy. Common adverse reactions include local injection site reactions, diarrhea, nausea, and fatigue.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
HIV-1 Infection
- The recommended dose of FUZEON is 90 mg (1 mL) twice daily injected subcutaneously into the upper arm, anterior thigh or abdomen. Each injection should be given at a site different from the preceding injection site, and only where there is no current injection site reaction from an earlier dose. FUZEON should not be injected near any anatomical areas where large nerves course close to the skin, such as near the elbow, knee, groin or the inferior or medial section of the buttocks, skin abnormalities, including directly over a blood vessel, into moles, scar tissue, bruises, or near the navel, surgical scars, tattoos or burn sites. Additional detailed information regarding the administration of FUZEON is described in the FUZEON Injection Instructions.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Enfuvirtide in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Enfuvirtide in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
HIV-1 Infection
- Insufficient data are available to establish a dose recommendation of FUZEON in pediatric patients below the age of 6 years. In pediatric patients 6 years through 16 years of age, the recommended dosage of FUZEON is 2 mg/kg twice daily up to a maximum dose of 90 mg twice daily injected subcutaneously into the upper arm, anterior thigh or abdomen. Each injection should be given at a site different from the preceding injection site and only where there is no current injection site reaction from an earlier dose. FUZEON should not be injected into moles, scar tissue, bruises or the navel. Table 1 contains dosing guidelines for FUZEON based on body weight. Weight should be monitored periodically and the FUZEON dose adjusted accordingly.

This image is provided by the National Library of Medicine.

Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Enfuvirtide in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Enfuvirtide in pediatric patients.
Contraindications
- FUZEON is contraindicated in patients with known hypersensitivity to FUZEON or any of its components.
Warnings
Precautions
- Local Injection Site Reactions (ISRs)
- The majority of subjects (98%) receiving FUZEON in randomized, controlled, open-label, multicenter clinical trials had at least one local injection site reaction; ISRs occurred throughout treatment with FUZEON. Manifestations may include pain and discomfort, induration, erythema, nodules and cysts, pruritus, and ecchymosis. Reactions are often present at more than one injection site. Patients must be familiar with the FUZEON Injection Instructions in order to know how to inject FUZEON appropriately and how to monitor carefully for signs or symptoms of cellulitis or local infection.
- Administration with Biojector® 2000
- Nerve pain (neuralgia and/or paresthesia) lasting up to 6 months associated with administration at anatomical sites where large nerves course close to the skin, bruising and hematomas have occurred with use of the Biojector 2000 needle-free device for administration of FUZEON. Patients receiving anticoagulants or persons with hemophilia, or other coagulation disorders, may have a higher risk of post-injection bleeding.
- Pneumonia
- An increased rate of bacterial pneumonia was observed in subjects treated with FUZEON in the Phase 3 clinical trials compared to the control arm. The incidence of pneumonia was 2.7% or 3.2 events/100 patient-years in subjects receiving FUZEON+background regimen. On analysis of all diagnoses of pneumonia (pneumonia, bacterial pneumonia, bronchopneumonia, and related terms) in T20-301 and T20-302, an increased rate of bacterial pneumonia was observed in subjects treated with FUZEON compared to the control arm (6.9%, 6.7 pneumonia events per 100 patient-years versus 0.6 events per 100 patient-years, respectively). Approximately half of the study subjects with pneumonia required hospitalization. Three subject deaths in the FUZEON arm were attributed to pneumonia; all three had serious concomitant AIDS-related illnesses that contributed to their deaths. Risk factors for pneumonia included low initial CD4 lymphocyte count, high initial viral load, intravenous drug use, smoking, and a prior history of lung disease.
- Because it was unclear whether the higher incidence rate of pneumonia was related to FUZEON use, an observational study in 1850 HIV-infected patients (740 FUZEON treated patients and 1110 non-FUZEON treated patients) was conducted to evaluate the risk of pneumonia in patients treated with FUZEON. A total of 123 patients had a confirmed or probable pneumonia event in this study (62 in the FUZEON treatment arm with 1962 patient-years of observation and 61 in the non-FUZEON treatment arm with 3378 patient-years of observation). The incidence of pneumonia was 3.2 events/100 patient-years in the FUZEON treatment arm and 1.8 events/100 patient-years in the non-FUZEON treatment arm. The hazard ratio, adjusting for other baseline risk factors, was 1.34 (95% C.I. = 0.90 – 2.00). Based on this observational study, it is not possible to exclude an increased risk of pneumonia in patients treated with FUZEON compared to non-FUZEON treated patients.
- It is unclear if the increased incidence of pneumonia is related to FUZEON use. However, because of these findings, patients with HIV-1 infection should be carefully monitored for signs and symptoms of pneumonia, especially if they have underlying conditions which may predispose them to pneumonia. Risk factors for pneumonia included low initial CD4 cell count, high initial viral load, intravenous drug use, smoking, and a prior history of lung disease.
- Hypersensitivity Reactions
- Systemic hypersensitivity reactions have been associated with FUZEON therapy and may recur on re-challenge. Hypersensitivity reactions have occurred in <1% of subjects studied and have included combinations of: rash, fever, nausea and vomiting, chills, rigors, hypotension, and/or elevated serum liver transaminases. Other adverse events that may be immune mediated and have been reported in subjects receiving FUZEON include primary immune complex reaction, respiratory distress, glomerulonephritis, and Guillain-Barre syndrome. Patients developing signs and symptoms suggestive of a systemic hypersensitivity reaction should discontinue FUZEON and should seek medical evaluation immediately. Therapy with FUZEON should not be restarted following systemic signs and symptoms consistent with a hypersensitivity reaction. Risk factors that may predict the occurrence or severity of hypersensitivity to FUZEON have not been identified.
- Non-HIV Infected Individuals
- There is a theoretical risk that FUZEON use may lead to the production of anti-enfuvirtide antibodies which cross react with HIV gp41. This could result in a false positive HIV test with an ELISA assay; a confirmatory western blot test would be expected to be negative. FUZEON has not been studied in non-HIV infected individuals.
- Immune Reconstitution Syndrome
- Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including FUZEON. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP] or tuberculosis), which may necessitate further evaluation and treatment.
- Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution, however, the time to onset is more variable, and can occur many months after initiation of treatment.
Adverse Reactions
Clinical Trials Experience
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
- The overall safety profile of FUZEON is based on 2131 subjects who received at least 1 dose of FUZEON during various clinical trials. This includes 2051 adults, 658 of whom received the recommended dose for greater than 48 weeks, and 63 pediatric subjects.
- Assessment of treatment-emergent adverse events is based on the pooled data from the two randomized, controlled, open-label, multicenter trials in treatment-experienced subjects, T20-301 (TORO 1) and T20-302 (TORO 2).
- Local Injection Site Reactions
- Local injection site reactions were the most frequent adverse events associated with the use of FUZEON. In T20-301 and T20-302, 98% of subjects had at least one local injection site reaction (ISR). A total of 7% of subjects discontinued treatment with FUZEON because of ISRs (4%) or difficulties with injecting FUZEON (3%) such as injection fatigue and inconvenience. Eighty-five percent of subjects experienced their first ISR during the initial week of treatment; ISRs continued to occur throughout treatment with FUZEON. For most subjects the severity of signs and symptoms associated with ISRs did not change during the 48 weeks of treatment. The majority of ISRs were associated with erythema, induration, the presence of nodules or cysts, and mild to moderate pain at the injection site (Table 2). In addition, the average duration of individual ISRs was between three and seven days in 41% of subjects and more than seven days in 24% of subjects. Also, the numbers of ISRs per subject at any one time was between six to 14 ISRs in 26% of subjects and more than 14 ISRs in 1.3% of subjects. Infection at the injection site (including abscess and cellulitis) was reported in 1.7% of adult subjects.

This image is provided by the National Library of Medicine.

- Other Adverse Events
- In T20-301 and T20-302, after study week 8, subjects on background alone who met protocol defined criteria for virological failure were permitted to revise their background regimens and add FUZEON. Exposure on FUZEON+background was 557 patient-years, and to background alone 162 patient-years. Due to this difference in exposure, safety results are expressed as the number of patients with an adverse event per 100 patient-years of exposure. For FUZEON+background, adverse events are also displayed by percent of subjects.
- The events most frequently reported in subjects receiving FUZEON+background regimen, excluding ISRs, were diarrhea (38 per 100 patient-years or 31.7%), nausea (27 per 100 patient-years or 22.8%), and fatigue (24 per 100 patient-years or 20.2%). These events were also commonly observed in subjects that received background regimen alone: diarrhea (73 per 100 patient-years), nausea (50 per 100 patient-years), and fatigue (38 per 100 patient-years).
- Treatment-emergent adverse events, regardless of causality and excluding ISRs, from Phase 3 studies are summarized for adult subjects, in Table 3. Any Grade 2 or above events occurring at ≥2 percent of subjects and at a higher rate in subjects treated with FUZEON are summarized in Table 3; events that occurred at a higher rate in the control arms are not displayed.
- Rates of adverse events for subjects who switched to FUZEON after virological failure were similar.

This image is provided by the National Library of Medicine.

- Less Common Events
- The following adverse events have been reported in 1 or more subjects; however, a causal relationship to FUZEON has not been established.
Immune System Disorders
Worsening abacavir hypersensitivity reaction
Renal and Urinary Disorders
Glomerulonephritis; tubular necrosis; renal insufficiency; renal failure (including fatal cases)
Blood and Lymphatic Disorders
Thrombocytopenia; neutropenia; fever; lymphadenopathy
Endocrine and Metabolic
Infections
Nervous System Disorders
Taste disturbance; Guillain-Barre syndrome (fatal); sixth nerve palsy; peripheral neuropathy
Cardiac Disorders
Unstable angina pectoris
Gastrointestinal Disorders
Constipation; abdominal pain upper
General
Hepatobiliary Disorders
Toxic hepatitis; hepatic steatosis
Investigations
Increased amylase; increased lipase; increased AST; increased GGT; increased triglycerides
Psychiatric Disorders
Insomnia; depression; anxiety; suicide attempt
Respiratory, Thoracic, and Mediastinal Disorders
Pneumopathy; respiratory distress; cough
Skin and Subcutaneous Tissue Disorders
- Adverse Events in Pediatric Patients
- FUZEON has been studied in 63 pediatric subjects 5 through 16 years of age with duration of FUZEON exposure ranging from 1 dose to 134 weeks. Adverse experiences seen during clinical trials were similar to those observed in adult subjects, although infections at site of injection (cellulitis or abscess) were more frequent in adolescents than in adults, with 4 events occurring in 3 of 28 (11%) subjects.
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Enfuvirtide in the drug label.
Drug Interactions
- Potential for FUZEON to Affect Other Drugs
- Based on the results from an in vitro human microsomal study, enfuvirtide is not an inhibitor of CYP450 enzymes. In an in vivo human metabolism study (N=12), FUZEON at the recommended dose of 90 mg twice daily did not alter the metabolism of CYP3A4, CYP2D6, CYP1A2, CYP2C19 or CYP2E1 substrates.
- Potential for Other Drugs to Affect Enfuvirtide
- Based on the available data, co-administration of FUZEON and other drugs which are inducers or inhibitors of CYP450 is not expected to alter the pharmacokinetics of enfuvirtide. No dose adjustments are needed when FUZEON is co-administered with other antiretroviral and non-antiretroviral drugs.
Use in Specific Populations
Pregnancy
- Pregnancy Category B
- Reproduction studies have been performed in rats and rabbits at doses up to 27 times and 3.2 times the adult human dose on a m2 basis and have revealed no evidence of impaired fertility or harm to the fetus due to enfuvirtide. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
- Antiretroviral Pregnancy Registry
- To monitor maternal-fetal outcomes of pregnant women exposed to FUZEON and other antiretroviral drugs, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Enfuvirtide in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Enfuvirtide during labor and delivery.
Nursing Mothers
- The Centers for Disease Control and Prevention recommends that HIV-infected mothers not breast-feed their infants to avoid the risk of postnatal transmission of HIV. It is not known whether enfuvirtide is excreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breast-feed if they are receiving FUZEON.
- Studies where radio-labeled 3H-enfuvirtide was administered to lactating rats indicated that radioactivity was present in the milk. It is not known whether the radioactivity in the milk was from radio-labeled enfuvirtide or from radio-labeled metabolites of enfuvirtide (i.e., amino acids and peptide fragments).
Pediatric Use
- The safety and pharmacokinetics of FUZEON have been evaluated in the age groups of 6 to 16 years of age supported by evidence from adequate and well-controlled studies of FUZEON in adults. Limited efficacy data are available in pediatric subjects 6 years of age and older.
- Sixty-three HIV-1 infected pediatric subjects ages 5 through 16 years have received FUZEON in two open-label, single-arm clinical trials. Adverse experiences, including ISRs, were similar to those observed in adult subjects.
- T20-204 was an open-label, multicenter trial that evaluated the safety and antiviral activity of FUZEON in treatment-experienced pediatric subjects. Eleven subjects from 6 to 12 years were enrolled (median age of 9 years). Median baseline CD4 cell count was 495 cells/µL and the median baseline HIV-1 RNA was 4.6 log10 copies/mL.
- Ten of the 11 study subjects completed 48 weeks of chronic therapy. At week 48, 6/11 (55%) subjects had ≥1 log10 decline in HIV-1 RNA and 4/11 (36%) subjects were below 400 copies/mL of HIV-1 RNA. The median changes from baseline (for the As Treated population) in HIV-1 RNA and CD4 cell count were -1.48 log10 copies/mL and +122 cells/µL, respectively.
- T20-310 was an open-label, multicenter trial that evaluated the pharmacokinetics, safety, and antiviral activity of FUZEON in treatment-experienced pediatric subjects and adolescents. Fifty-two subjects from 5 through 16 years were enrolled (median age of 12 years). Median baseline CD4 cell count was 117 cells/µL and the median baseline HIV-1 RNA was 5.0 log10 copies/mL.
- Thirty-two of the 52 study subjects completed 48 weeks of chronic therapy. At week 48, 17/52 (33%) of subjects had ≥1 log10 decline in HIV-1 RNA, 11/52 (21%) of subjects were below 400 copies/mL of HIV-1 RNA and 5/52 (10%) were below 50 copies/mL. The median changes from baseline (for the As Treated population) in HIV-1 RNA and CD4 cell count were -1.17 log10 copies/mL and +106 cells/µL, respectively.
Geriatic Use
- Clinical studies of FUZEON did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, appropriate caution should be exercised in the administration and monitoring of FUZEON in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Enfuvirtide with respect to specific gender populations.
Race
There is no FDA guidance on the use of Enfuvirtide with respect to specific racial populations.
Renal Impairment
- No dose adjustments of enfuvirtide are needed in patients with renal impairment.
Hepatic Impairment
- No dose adjustments of enfuvirtide are needed in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Enfuvirtide in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Enfuvirtide in patients who are immunocompromised.
Administration and Monitoring
Administration
- Subcutaneous
Monitoring
There is limited information regarding Monitoring of Enfuvirtide in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Enfuvirtide in the drug label.
Overdosage
Chronic Overdose
There is limited information regarding Chronic Overdose of Enfuvirtide in the drug label.
Pharmacology
Enfuvirtide
| |
| Systematic (IUPAC) name | |
| ? | |
| Identifiers | |
| CAS number | |
| ATC code | J05 |
| PubChem | |
| DrugBank | |
| Chemical data | |
| Formula | Template:OrganicBox atomTemplate:OrganicBox atomTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBox atomTemplate:OrganicBoxTemplate:OrganicBox atomTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBox |
| Mol. mass | 4492.1 g/mol |
| SMILES | & |
| Pharmacokinetic data | |
| Bioavailability | 84.3% (SC) |
| Protein binding | 92% |
| Metabolism | Hepatic |
| Half life | 3.8 hours |
| Excretion | unknown |
| Therapeutic considerations | |
| Pregnancy cat. | |
| Legal status |
Prescription Only (S4)(AU) ?(CA) POM(UK) [[Prescription drug|Template:Unicode-only]](US) S4 |
| Routes | Subcutaneous (SC) |
Mechanism of Action
- Enfuvirtide interferes with the entry of HIV-1 into cells by inhibiting fusion of viral and cellular membranes. Enfuvirtide binds to the first heptad-repeat (HR1) in the gp41 subunit of the viral envelope glycoprotein and prevents the conformational changes required for the fusion of viral and cellular membranes.
Structure
- FUZEON (enfuvirtide) is an inhibitor of the fusion of HIV-1 with CD4 cells. Enfuvirtide is a linear 36-amino acid synthetic peptide with the N-terminus acetylated and the C-terminus is a carboxamide. It is composed of naturally occurring L-amino acid residues.
- Enfuvirtide is a white to off-white amorphous solid. It has negligible solubility in pure water and the solubility increases in aqueous buffers (pH 7.5) to 85-142 g/100 mL. The empirical formula of enfuvirtide is C204H301N51O64, and the molecular weight is 4492. It has the following primary amino acid sequence:
- CH3CO-Tyr-Thr-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lys-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Phe-NH2 and the following structural formula:

This image is provided by the National Library of Medicine.

- The drug product, FUZEON (enfuvirtide) for Injection, is a white to off-white, sterile, lyophilized powder. Each single-use vial contains 108 mg of enfuvirtide for the delivery of 90 mg. Prior to subcutaneous administration, the contents of the vial are reconstituted with 1 mL of Sterile Water for Injection to provide the delivery of 1 mL of the solution. Each 1 mL of the reconstituted solution contains approximately 90 mg of enfuvirtide with approximate amounts of the following excipients: 22.55 mg of mannitol, 2.39 mg of sodium carbonate (anhydrous), and sodium hydroxide and hydrochloric acid for pH adjustment as needed. The reconstituted solution has an approximate pH of 9.0.
Pharmacodynamics
There is limited information regarding Pharmacodynamics of Enfuvirtide in the drug label.
Pharmacokinetics
- The pharmacokinetic properties of enfuvirtide were evaluated in HIV-1 infected adult and pediatric subjects.
- Absorption
- Following a 90-mg single subcutaneous injection of FUZEON into the abdomen in 12 HIV-1 infected subjects, the mean (±SD) Cmax was 4.59 ± 1.5 µg/mL, AUC was 55.8 ± 12.1 µg∙h/mL and the median Tmax was 8 hours (ranged from 3 to 12 h). The absolute bioavailability (using a 90-mg intravenous dose as a reference) was 84.3% ± 15.5%. Following 90-mg twice daily dosing of FUZEON subcutaneously in combination with other antiretroviral agents in 11 HIV-1 infected subjects, the mean (±SD) steady-state Cmax was 5.0 ± 1.7 µg/mL, Ctrough was 3.3 ± 1.6 µg/mL, AUC0-12h was 48.7 ± 19.1 µg∙h/mL, and the median Tmax was 4 hours (ranged from 4 to 8 h).
- Absorption of the 90-mg dose was comparable when injected into the subcutaneous tissue of the abdomen, thigh or arm.
- Distribution
- The mean (±SD) steady-state volume of distribution after intravenous administration of a 90-mg dose of FUZEON (N=12) was 5.5 ± 1.1 L.
- Enfuvirtide is approximately 92% bound to plasma proteins in HIV-infected plasma over a concentration range of 2 to 10 µg/mL. It is bound predominantly to albumin and to a lower extent to α-1 acid glycoprotein.
- The CSF levels of enfuvirtide (measured from 2 hours to 18 hours after administration of enfuvirtide) in 4 HIV-infected subjects were below the limit of quantification (0.025 µg/mL).
- Metabolism/Elimination
- As a peptide, enfuvirtide is expected to undergo catabolism to its constituent amino acids, with subsequent recycling of the amino acids in the body pool.
- Mass balance studies to determine elimination pathway(s) of enfuvirtide have not been performed in humans.
- In vitro studies with human microsomes and hepatocytes indicate that enfuvirtide undergoes hydrolysis to form a deamidated metabolite at the C-terminal phenylalanine residue, M3. The hydrolysis reaction is not NADPH dependent. The M3 metabolite is detected in human plasma following administration of enfuvirtide, with an AUC ranging from 2.4% to 15% of the enfuvirtide AUC.
- Following a 90-mg single subcutaneous dose of enfuvirtide (N=12) the mean ±SD elimination half-life of enfuvirtide is 3.8 ± 0.6 h and the mean ±SD apparent clearance was 24.8 ± 4.1 mL/h/kg. Following 90-mg twice daily dosing of FUZEON subcutaneously in combination with other antiretroviral agents in 11 HIV-1 infected subjects, the mean ±SD apparent clearance was 30.6 ± 10.6 mL/h/kg.
- Special Populations
- Hepatic Impairment
- Formal pharmacokinetic studies of enfuvirtide have not been conducted in subjects with hepatic impairment.
- Renal Impairment
- Analysis of plasma concentration data from subjects in clinical trials indicated that the clearance of enfuvirtide is not affected in patients with creatinine clearance greater than 35 mL/min. The results of a renal impairment study indicate clearance of enfuvirtide was reduced by 38% in subjects with severe renal impairment (CL = 11 – 35 mL/min; n = 4) and by 14 - 28% in subjects with end-stage renal disease maintained on dialysis (n = 8) compared to subjects with normal renal function (CL >80 mL/min; n = 8). Hemodialysis did not significantly alter enfuvirtide clearance.
- No dose adjustment is recommended for patients with impaired renal function.
- Gender and Weight
- Analysis of plasma concentration data from subjects in clinical trials indicated that the clearance of enfuvirtide is 20% lower in females than males after adjusting for body weight.
- Enfuvirtide clearance decreases with decreased body weight irrespective of gender. Relative to the clearance of a 70-kg male, a 40-kg male will have 20% lower clearance and a 110-kg male will have a 26% higher clearance. Relative to a 70-kg male, a 40-kg female will have a 36% lower clearance and a 110-kg female will have the same clearance.
- No dose adjustment is recommended for weight or gender.
- Race
- Analysis of plasma concentration data from subjects in clinical trials indicated that the clearance of enfuvirtide was not different in Blacks compared to Caucasians. Other pharmacokinetic studies suggest no difference between Asians and Caucasians after adjusting for body weight.
- Pediatric Patients
- The pharmacokinetics of enfuvirtide have been studied in 23 pediatric subjects aged 6 through 16 years at a dose of 2 mg/kg. Enfuvirtide pharmacokinetics were determined in the presence of concomitant medications including antiretroviral agents. A dose of 2 mg/kg twice daily (maximum 90 mg twice daily) provided enfuvirtide plasma concentrations similar to those obtained in adult subjects receiving 90 mg twice daily.
- In the 23 pediatric subjects receiving the 2 mg/kg twice daily dose, the mean ±SD steady-state AUC was 56.3 ± 22.3 µg∙h/mL, Cmax was 6.3 ± 2.4 µg/mL, Ctrough was 3.1 ± 1.5 µg/mL, and apparent clearance was 40 ± 17 mL/h/kg.
- Geriatric Patients
- The pharmacokinetics of enfuvirtide have not been studied in patients over 65 years of age.
- Table 5 shows the results of the drug-drug interaction studies conducted between FUZEON and the following drugs: ritonavir, saquinavir/ritonavir, and rifampin.

This image is provided by the National Library of Medicine.

Nonclinical Toxicology
- Carcinogenesis
- Long-term animal carcinogenicity studies of enfuvirtide have not been conducted.
- Mutagenesis
- Enfuvirtide was neither mutagenic nor clastogenic in a series of in vivo and in vitro assays including the Ames bacterial reverse mutation assay, a mammalian cell forward gene mutation assay in AS52 Chinese Hamster ovary cells or an in vivo mouse micronucleus assay.
- Impairment of Fertility
- Enfuvirtide produced no adverse effects on fertility in male or female rats at doses up to 1.6 times the maximum recommended adult human daily dose on a m2 basis.
Clinical Studies
Studies in Antiretroviral Experienced Patients
- T20-301 and T20-302 were randomized, controlled, open-label, multicenter trials in HIV-1 infected subjects. Subjects were required to have either (1) viremia despite 3 to 6 months prior therapy with a nucleoside reverse transcriptase inhibitor (NRTI), non-nucleoside reverse transcriptase inhibitor (NNRTI), and protease inhibitor (PI) or (2) viremia and documented resistance or intolerance to at least one member in each of the NRTI, NNRTI, and PI classes.
- All subjects received an individualized background regimen consisting of 3 to 5 antiretroviral agents selected on the basis of the subject's prior treatment history and baseline genotypic and phenotypic viral resistance measurements. Subjects were then randomized at a 2:1 ratio to FUZEON 90 mg twice daily with background regimen or background regimen alone.
- After week 8, subjects on either treatment arm who met protocol defined criteria for virological failure were permitted to revise their background regimens; those on background regimen alone were also permitted to add FUZEON.
- Demographic characteristics for studies T20-301 and T20-302 are shown in Table 6. Subjects had prior exposure to a median of 12 antiretrovirals for a median of 7 years.

This image is provided by the National Library of Medicine.

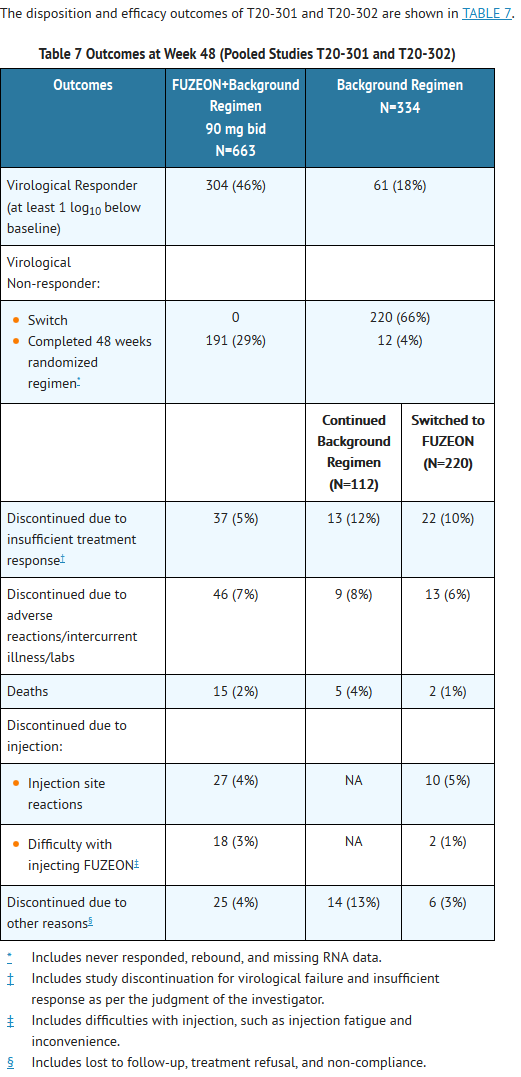
This image is provided by the National Library of Medicine.

- At 48 weeks, 154 (23%) of subjects in the FUZEON+background regimen and 27 (8%) in the background regimen alone had HIV-1 RNA levels <50 copies/mL, and 225 (34%) of subjects receiving FUZEON+background regimen had HIV-1 RNA levels <400 copies/mL compared to 44 (13%) in the background regimen alone. Subjects achieving HIV-1 RNA levels <50 copies/mL were included in the <400 copies/mL category and both categories were incorporated in the overall virologic responder category of achieving HIV-1 RNA at least 1 log10 below baseline.
- The mean log change in HIV-1 RNA from baseline was -1.4 log10 copies/mL in subjects receiving FUZEON+background and -0.5 in those receiving background alone. The mean change in CD4 cell count from baseline to week 48 was +91 cells/mm3 in the FUZEON+background arm and +45 cells/mm3 in the background alone arm.
- Subjects in the FUZEON+background arm achieved a better virologic and immunologic outcome than subjects in the background alone arm across all subgroups based on baseline CD4 cell count, baseline HIV-1 RNA, number of prior ARVs or number of active ARVs in the background regimen.
How Supplied
- FUZEON (enfuvirtide) for Injection is a white to off-white, sterile, lyophilized powder and it is packaged in a single-use clear glass vial containing 108 mg of enfuvirtide for the delivery of approximately 90 mg/1 mL when reconstituted with 1 mL of Sterile Water for Injection.
- FUZEON is available in a Convenience Kit containing 60 single-use vials of FUZEON (90 mg strength), 60 vials (2 cartons of 30 each) of Sterile Water for Injection (1 mL per vial), 60 reconstitution syringes (3 cc), 60 administration syringes (1 cc), Package Insert, Patient Package Insert, and Injection Instructions (NDC 0004-0381-40).
- Storage Conditions
- Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F).
- Reconstituted solution should be stored in the original vial under refrigeration at 2° to 8°C (36° to 46°F) and used within 24 hours.
Storage
There is limited information regarding Enfuvirtide Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Enfuvirtide |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Enfuvirtide |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- To assure safe and effective use of FUZEON, the following information and instructions should be given to patients:
- Patients should be informed that injection site reactions occur in almost all patients taking FUZEON. Patients must be familiar with the FUZEON Injection Instructions for instructions on how to appropriately inject FUZEON and how to carefully monitor for signs or symptoms of cellulitis or local infection. Patients should be instructed when to contact their healthcare provider about these reactions.
- Patients should be made aware that an increased rate of bacterial pneumonia was observed in subjects treated with FUZEON in clinical trials. Patients should be advised to seek medical evaluation immediately if they develop signs or symptoms suggestive of pneumonia (cough with fever, rapid breathing, shortness of breath).
- Patients should be advised of the possibility of a systemic hypersensitivity reaction to FUZEON. Patients should be advised to discontinue therapy and immediately seek medical evaluation if they develop signs/symptoms of systemic hypersensitivity such as combinations of rash, fever, nausea and vomiting, chills, rigors, and/or hypotension.
- FUZEON is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using FUZEON.
- Patients should be advised to avoid doing things that can spread HIV-1 infection to others.
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
- Do not breastfeed. We do not know if FUZEON can be passed to your baby in your breast milk and whether it could harm your baby. Also, mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.
- FUZEON must be taken as part of a combination antiretroviral regimen. Use of FUZEON alone may lead to rapid development of virus resistant to FUZEON and possibly other agents of the same class.
- Patients and caregivers must be instructed in the use of aseptic technique when administering FUZEON in order to avoid injection site infections. Appropriate training for FUZEON reconstitution and self-injection must be given by a healthcare provider, including a careful review of the FUZEON Patient Package Insert and FUZEON Injection Instructions. The first injection should be performed under the supervision of an appropriately qualified healthcare provider. It is recommended that the patient and/or caregiver's understanding and use of aseptic injection techniques and procedures be periodically re-evaluated.
- Patients and caregivers should be instructed on the preferred anatomical sites for administration (upper arm, abdomen, anterior thigh). FUZEON should not be injected near any anatomical areas where large nerves course close to the skin, such as near the elbow, knee, groin or the inferior or medial sections of the buttocks, skin abnormalities, including directly over a blood vessel, into moles, scar tissue, bruises, or near the navel, surgical scars, tattoos or burn sites.
- Patients and caregivers should be instructed in the proper techniques for preparation, injection and disposal of needles and syringes (including not recapping needles) in order to avoid needle stick injuries. Patients should be told not to reuse needles or syringes, and be instructed in safe disposal procedures including the use of a puncture-resistant container for disposal of used needles and syringes. Patients must be instructed on the safe disposal of full containers as per local requirements. Caregivers who experience an accidental needle stick after patient injection should contact a healthcare provider immediately.
- Patients should contact their healthcare provider for any questions regarding the administration of FUZEON.
- Patients should inform their healthcare provider if they are pregnant, plan to become pregnant or become pregnant while taking this medication.
- Patients should inform their healthcare provider if they are breast-feeding.
- Patients should not change the dose or dosing schedule of FUZEON or any antiretroviral medication without consulting their healthcare provider.
- Patients should contact their healthcare provider immediately if they stop taking FUZEON or any other drug in their antiretroviral regimen.
- Patients should be told that they can obtain more information on the self-administration of FUZEON at www.FUZEON.com or by calling 1-877-4-FUZEON (1-877-438-9366).
- Patients should be advised that no studies have been conducted on the ability to drive or operate machinery while taking FUZEON. If patients experience dizziness while taking FUZEON, they should be advised to talk to their healthcare provider before driving or operating machinery.

This image is provided by the National Library of Medicine.

Precautions with Alcohol
- Alcohol-Enfuvirtide interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- FUZEON®[1]
Look-Alike Drug Names
There is limited information regarding Enfuvirtide Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Enfuvirtide
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Enfuvirtide |Label Name=Enfuvirtide09.png
}}
{{#subobject:
|Label Page=Enfuvirtide |Label Name=Enfuvirtide10.png
}}