Primitive neuroectodermal tumor (also known as "PNET") is a rare type of malignant neural crest tumor. PNET arises from the neuroectoderm, which is normally involved in the development of the nervous system. Apart from central nervous system (CNS), PNETs can involve other tissues originating from the neuroectoderm such as muscles and bones. PNET was first discovered by James Ewing, an American pathologist, in 1921. However, the term PNETs is more commonly was described in 1973 by Hart and Earle. In fact, PNETs are members of the Ewing tumor family. These tumors have small round cells, are believed to originate from postganglionic parasympathetic primordial cells and have mutations of the EWS gene. Due to their origin, PNETs can be found at any site within the parasympathetic system. Apart from Ewing Sarcoma (ES) and PNET, this family of tumors includes other tumors such as Askin's tumor (a malignant small-cell tumor in the chest) and paravertebral small-cell tumors. PNETs are divided into peripheral and central based on their presentation site. Central PNETs are more commonly seen among children and young adults and account for approximately 1% of PNETs. Peripheral PNETs mostly occur in bones and surrounding tissues. PNETs are more commonly seen among children and young adults. The median age at diagnosis is 25 years of age. PNETs are highly malignant and their prognosis is generally poor, however, the prognosis is more favorable for adult patients with PNET. The 5-survival rate of patients with PNET is less than 35%. The disease affects both men and women, however, there is a slight tendency toward affecting males in the cases of peripheral PNET.
Historical Perspective
Primitive neuroectodermal tumor was first discovered by James Ewing, an American pathologist, in 1921.[1]
Primitive neuroectodermal tumor may be classified based on the embryonic derivatives according to World Health Organization into 2 subtypes:[3][4]
central primitive neuroectodermal tumors(PNETs) which can include CNS neuroblastoma, CNS ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma.
Peripheral primitive neuroectodermal tumors (pPNETs), with most of the tumors arising in bone and surrounding tissues, especially of the limbs, paravertebral regions, chest (Askin's tumor), and pelvis.
Pathophysiology
The pathogenesis of peripheral primitive neuroectodermal tumor is characterized by the chromosomal translocation t(11;22)q24q12.[5][6]
This translocation fuses the EWS gene on chromosome 22 with the FLI1 gene on chromosome 11.
The EWS-FLI1 gene has been associated with the development of PNET involving the synthesis of adrenal pathway.
On gross pathology, white, hemorrhagic and necrotic mass are characteristic of PNET.[7]
On microscopy histopathological analysis, small round blue cells, fine chromatin, eosinophilic cytoplasm,homer-Wright rosettes, and high mitotic figures.[8][9]
Causes
The t (11;22)(q24;q12) translocation seen in most of the PNET cases seems to play a causative role.
Differentiating Primitive Neuroectodermal Tumor from Other Diseases
Primitive neuroectodermal tumor must be differentiated from other diseases that cause seizures or increase on intracranial pressure, such as astrocytoma, ependymoma, oligodendroglioma, intracranial teratoma, meningitis, encephalitis, and other brain tumors.
Epidemiology and Demographics
The prevalence of primitive neuroectodermal tumors remains unknown.
The annual incidence of PNETs from birth to 20 years of age is 2.9 per 1,000,000.
Primitive neuroectodermal tumor account for 4-17% of all soft tissue pediatric tumors.
The median age at diagnosis depends on the type of PNET and their location.
PNETs are more common among children and account for 2.5% of brain tumors in children [10].
Adults are much less affected by PNETs and these tumors account for 0.46% of adult brain tumors.
PNETs have a slight tendency toward affecting men compared to women [11].
PNETs are extremely rare in African and Asian individuals.
PNETs usually affect Hispanic and white individuals.
Peripheral PNET, has a tendency toward affecting Caucasians.
Risk Factors
Prenatal exposure to alcohol seems to be a risk factor for developing PNET [12].
Children who had lived in farms for at least 1 year showed an increased risk for PNET.
The majority of patients with primitive neuroectodermal tumor remain asymptomatic for years.
Early clinical features are often unspecific.
If left untreated, patients with primitive neuroectodermal tumor may progress to develop metastases.
Common complications of primitive neuroectodermal tumor, include increased intracranial pressure, cranial nerve palsy, and seizures.
Prognosis is similar for peripheral PNETs and central PNETs.
Prognosis is generally poor, and the 5-survival rate of patients with PNET less than 35% in adults and 64% in children[13].
Prognosis is more favorable for adult patients.
Tumors expressing CD99 are less aggressive after surgical resection and have better prognosis.
Features associated with good prognosis include early diagnosis, combinatorial treatmrnt approach including tumor resection, chemotherapy and radiotherapy, intratumoral calcification, Ki-67 <30%, high LDH, tumor volume >100 cc, and axial location.
Diagnosis
Symptoms
Clinical presentation of primitive neuroectodermal tumors is often non-specific and depend on the site of the tumor.
Microscopic features of PNETs show a proliferation as the mechanism of growth rather than infiltration.
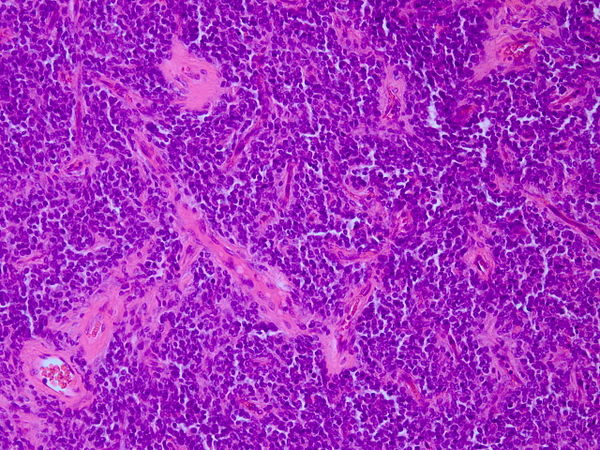
On microscopic histopathological analysis, characteristic findings of primitive neuroectodermal tumor, include:
Small blue cell tumorH&E staining of PNET. Courtesy of image: WikipediaRound hyperchromatic cells
Abundant mitotic figures
Homer-Wright rosettes, in which tumor cells surround neutrophils.
Fibrosis
short and round or spindle-shaped nuclei Immunohistochemical analysis can reveal differentiation toward different directions such as glial, neuronal and ependymal[14] .
Immunohistochemicsl analysis can be positive for CD99, CD56, Neuron-specific enolase (NSE), S-100 protein, synaptophysin, and chromogranin A.
Imaging Findings
MRI is the imaging modality of choice for primitive neuroectodermal tumor.
On CT, findings of primitive neuroectodermal tumor, may include:
Often seen as a large irregular mass
Typically iso to hyper-attenuating on non contrast imaging
Cystic components are common (65%)
Calcification can be common (70% )
Shows heterogenous contrast enhancement
On MRI, findings of primitive neuroectodermal tumor, may include:
T1: highly variable and can be hypo-intense to isointense, but usually hypo-intense
T2: generally high signal solid components
MRI with contrast shows acid enhancement
Cystic components and necrosis are common
Calcification and hemorrhage is common within the tumors
Tumor has well-defined borders without peripheral edema
T1 C+ (Gd): shows markedly heterogenous enhancement and leptomeningeal seeding is common
DWI: often shows restricted diffusion and solid composition in addition to enhancement which shows high vascularization of the tumor.
MR spectroscopy: elevated choline, decreased N-acetylaspartate (NAA), elevated taurine (Tau) peak (relatively specific for PNET).
In cases of peripheral PNET, whole body radioisotope scan can reveal the site of the tumor and possible metastases.
Treatment
Medical Therapy
There is no consensus in treatment of PNET.
Chemotherapy is controversial in treatment of PNET.
Temozolomide can be added to conventional treatment of excision and radiotherapy.
Surgery
Based on the site of the tumor, maximum resection must be performed.
Radiotherapy
7 to 8 weeks of radiotherapy at a dose of 50-55 Gy is recommended [15].
Prevention
There are no primary preventive measures available for primitive neuroectodermal tumor.
↑Chao, Xiaopei; Bi, Yalan; Li, Lei (2019). "Ovarian primary primitive neuroectodermal tumor: a review of cases at PUMCH and in the published literature". Orphanet Journal of Rare Diseases. 14 (1). doi:10.1186/s13023-019-1106-5. ISSN1750-1172.
↑Louis, David N.; Ohgaki, Hiroko; Wiestler, Otmar D.; Cavenee, Webster K.; Burger, Peter C.; Jouvet, Anne; Scheithauer, Bernd W.; Kleihues, Paul (2007). "The 2007 WHO Classification of Tumours of the Central Nervous System". Acta Neuropathologica. 114 (2): 97–109. doi:10.1007/s00401-007-0243-4. ISSN0001-6322.
↑Visee S, Soltner C, Rialland X, Machet MC, Loussouarn D, Milinkevitch S; et al. (2005). "Supratentorial primitive neuroectodermal
tumors of the brain: multidirectional differentiation
does not influence prognosis. A clinicopathological report of 18 patients". Histopathology. line feed character in |title= at position 41 (help)CS1 maint: Explicit use of et al. (link) CS1 maint: Multiple names: authors list (link)
↑Ohba S, Yoshida K, Hirose Y, Ikeda E, Kawase T. (2008). "A supratentorial primitive neuroectodermal tumor in an adult: a case report and review of the literature". J Neurooncol.CS1 maint: Multiple names: authors list (link)
↑Smoll NR. (2012). "Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs)". Cancer.
↑Pigott TJ, Punt JA, Lowe JS, Henderson MJ, Beck A, Gray T (1990). "The clinical, radiological and histopathological features of cerebral primitive neuroectodermal tumours". Br J Neurosurg.CS1 maint: Multiple names: authors list (link)
↑Batsakis JG, Mackay B, el-Naggar AK (1996). "Ewing's sarcoma and peripheral primitive neuroectodermal tumor: an interim report". Ann Otol Rhinol Laryngol.CS1 maint: Multiple names: authors list (link)