Ehlers-Danlos syndrome
For the WikiPatient page for this topic, click here
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Cafer Zorkun, M.D., Ph.D. [2]
Overview
Ehlers-Danlos syndrome is a group of rare genetic disorders affecting humans and domestic animals caused by a defect in collagen synthesis. Depending on the individual mutation, the severity of the disease can vary from mild to life-threatening. There is no known cure. Treatment is supportive.
Historical Perspective
The disease is named after two doctors, Edward Ehlers of Denmark, and Henri-Alexandre Danlos of France, who identified it at the turn of the 20th century. [1]
Classification
In the past, there were more than 10 recognized types of Ehlers-Danlos syndrome. In 1997, researchers proposed a simpler classification that reduced the number of major types to six and gave them descriptive names.[2] These six major types are listed below. Other types of the condition may exist, but they have been reported only in single families or are not well characterized. Except for hypermobility, the specific mutations involved have been identified and they can be precisely identified by genetic testing; this is valuable due to a great deal of variation in individual presentation of symptoms which may confuse classification of individuals on purely symptomatic basis. In order of prevalence in the population, they are:
| Name | Number | Description | OMIM | Gene(s) |
| Hypermobility | type 3 | Affects 1 in 10,000 to 15,000 and is caused by an autosomal dominant mechanism. Mutations in either of two separate genes (which are also involved in Vascular EDS and Tenascin-X deficiency EDS, respectively) may lead to this variant; it is the only type of EDS that cannot be diagnosed through skin / tissue samples but is rather diagnosed through use of clinical observations. Symptoms can include easy bruising, velvety-smooth skin, mildy hyperextensible skin, and loose, unstable joints. Joint dislocations and subluxations are common. Degenerative joint disease can occur; the pain associated with this condition is a serious complication. Unfortunately, pain medications are frequently underprescribed. Some individuals have mitral valve prolapse, which creates an increased risk for infective endocarditis during surgery, particularly dental surgery, as well as possibly progressing to a life-threatening degree of severity of the prognosis of mitral valve prolapse. | 130020 | COL3A1, TNXB |
| Classical | types 1 and 2 | Affects approximately 2 to 5 in 100,000 people. In addition to the joint and cardiac effects noted above for hypermobility, this variant is characterized by soft, highly elastic, velvety skin which may tear, bruise, or scar easily and/or be slow to heal, and which has a tendency to develop benign fatty growths as well as benign fibrous growths on pressure areas. Pregnancy can be life-threatening in this variant. It affects type-V collagen, as well as type I. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Vascular | type 4 | Is an autosomal dominant defect in the type 3 collagen synthesis; affecting approximately 1 in 100,000 people, it is clinically serious, reducing life expectancy to around 40 years. Joint symptoms are usually limited to the fingers or toes, but the delicate skin noted above is joined by fragile blood vessel walls and organ membranes, with a tendency to rupture or develop aneurysms. Thin, translucent skin, Arterial/intestinal/uterine fragility or rupture, Extensive bruising, Characteristic facial appearance - Distinctive facial features are associated with this variant; large eyes, small chin, thin nose and lips, and may also have lobeless (attached) ears, midline (betwen the eyes) facial flattening. Along with the above characteristics, a majority have reported that they sleep with their eyes half (partially) open. Pregnancy can be life-threatening in this variant as well. | 130050 | COL3A1 |
| Kyphoscoliosis | type 6 | Is an autosomal recessive defect due to deficiency of an enzyme called lysyl hydroxylase; it is very rare, with fewer than 60 cases reported. Symptoms include progressive scoliosis, progressive severe weakness of muscles, and fragile sclera. | 225400, 229200 | PLOD1 |
| Arthrochalasis | types 7A and 7B | Is also very rare, with about 30 cases reported. This variant may result in very loose and unstable joints, including the hips, which may lead to early and/or severe osteoarthritis and fractures, and stretchy, fragile skin. It affects type-I collagen. | 130060 | COL1A1, COL1A2 |
| Dermatosparaxis | type 7C | Also very rare, with about 10 cases reported. This variant combines the loose and unstable joints with extremely fragile skin which loses elasticity. | 225410 | ADAMTS2 |
It is very important to note that while the above symptomatology is clean and defined the disease itself rarely obeys these neat categorizations. Cross-over symptoms for all types are prevalent and lead to under-diagnosis or mis-diagnosis. No patient should assume or rely on the "fact" they have a certain type of EDS when cross-over symptoms are evident and can be life-threatening.
"The large number of distinct types of the Ehlers-Danlos syndrome that have already been identified indicates great heterogeneity, but clearly that heterogeneity is not exhausted by the present classification." [3] Examples of types of related syndromes other than those above reported in the medical literature include:
- 305200 -- Type 5
- 130080 -- Type 8 - unspecified gene, locus 12p13
- 225310 -- Type 10 - unspecified gene, locus 2q34
- 608763 -- Beasley-Cohen type
- 130070 -- Progeroid form - B4GALT7
- 606408 -- Due to Tenascin-X deficiency - TNXB
- 130090 -- Type unspecified
Pathophysiology
Genetics
Mutations in the ADAMTS2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, PLOD1 and TNXB genes cause Ehlers-Danlos syndrome.
Mutations in these genes usually alter the structure, production, or processing of collagen or proteins that interact with collagen. Collagen provides structure and strength to connective tissue throughout the body. A defect in collagen can weaken connective tissue in the skin, bones, blood vessels, and organs, resulting in the features of the disorder.
Inheritance patterns depend on the type of Ehlers-Danlos syndrome. Most forms of the condition are inherited in an autosomal dominant pattern, which means only one of the two copies of the gene in question must be altered to cause the disorder. The minority are inherited in an autosomal recessive pattern, which means both copies of the gene must be altered for a person to be affected by the condition. Please refer to the summary for each type of Ehlers-Danlos syndrome for a discussion of its inheritance pattern.
Epidemiology and Demographics
The overall prevalence of all types of Ehlers-Danlos syndrome may be about 1 in 5,000 births worldwide. The prevalence of the six types differs dramatically. The most common are the hypermobile forms (the classical and hypermobility types). Other forms are very rare. For example, fewer than 10 infants and children with the dermatosparaxis type have been described worldwide. It affects both males and females of all racial and ethnic backgrounds.
Diagnosis
Symptoms
Symptoms vary widely based on which type of Ehlers-Danlos Syndrome (EDS) the patient has. In each case, however, the symptoms are ultimately due to faulty or reduced amounts of collagen. For example, in the most common type of EDS, Hypermobility Type, symptoms often include unstable, flexible joints with a painful tendency to dislocate and subluxate. This is due to ligaments which, because they are lacking proper collagen--the molecule that provides strength to ligaments--are overly stretchable. The so-called Classic EDS Type features skin that forms cigarette-paper-like scars. Another type of collagen is usually responsible for lending strength to skin (and scars). The most serious type of EDS, Vascular EDS, can result in premature death via vascular (blood vessel) and organ rupture. [3] Again, another type collagen is necessary to give strength to the walls of blood vessels and the walls of hollow organs (such as the large bowel, aka colon). (It should be noted that Vascular EDS is also one of the most rare types of the disease.) See table below for a more extensive list of symptoms for each type of EDS. You will see some cross-over or similarity of symptoms among the various types. For instance, many of the types feature velvety or hyperextensible skin. In addition, persons with Hypermobility Type often have very stretchy ligements (leading to frequent subluxations/dislocations) while those with Vascular Type have ligaments that rupture.
Physical examination
-
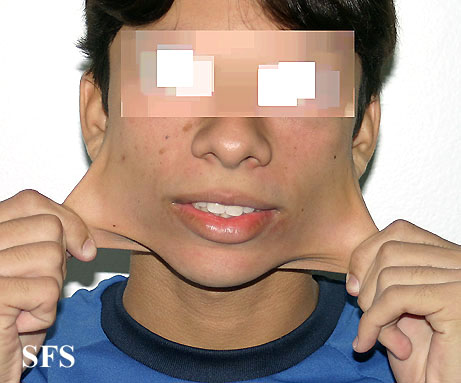 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
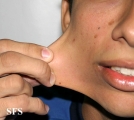 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
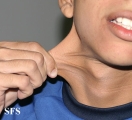 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
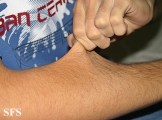 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
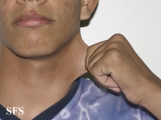 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
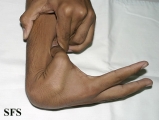 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
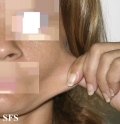 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
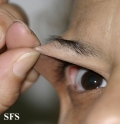 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
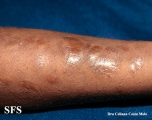 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
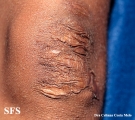 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
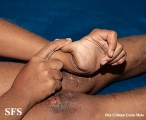 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas -
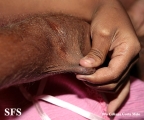 Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Adapted from Dermatology Atlas














See also
References
- ↑ "Uncovered: How U.S. Health System Can Fail Even the Insured - A Woman Endures 16-Month Odyssey To Get a Diagnosis", John Carreyrou, Wall Street Journal, November 16, 2007
- ↑ Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ (1998). "Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK)". Am J Med Genet. 77 (1): 31–7. PMID 9557891.
- ↑ Lawrence EJ (2005). "The clinical presentation of Ehlers-Danlos syndrome". Adv Neonatal Care. 5 (6): 301–14. PMID 16338669.
External links
- EDS Today - The Newsletter for, by, and about people with Ehlers Danlos Syndrome (EDS)
- Ehlers-Danlos Network C.A.R.E.S. INC - Children, Adults, Research, Education, Support
- Ehlers-Danlos Support Group
- Ehlers-Danlos National Foundation
- Ehlers Danlos Foundation Of New Zealand
- eds at NIH/UW GeneTests
de:Ehlers-Danlos-Syndrom nl:Syndroom van Ehlers-Danlos sr:Елерс-Данлос синдром fi:Ehlers-Danlosin syndrooma sv:Ehlers-Danlos syndrom