Wilson's disease pathophysiology: Difference between revisions
No edit summary |
No edit summary |
||
| (15 intermediate revisions by one other user not shown) | |||
| Line 10: | Line 10: | ||
{{CMG}}; {{AE}} {{AEL}} | {{CMG}}; {{AE}} {{AEL}} | ||
==Overview== | ==Overview== | ||
[[Copper]] is one of the essential elements for the [[human body]]. Copper must be absorbed and transported in order to function properly. Copper is transported bound to [[metallothionein]] or carried by [[ATOX1]] to the [[Golgi apparatus|trans-Golgi network]]. Impairment of copper incorportation in formation of [[ceruloplasmin]] will lead to increase the serum level of the [[copper]]. The increase of copper level will lead to accumulation of the excess amount in the [[liver]] and other organs. [[ATP7B]] [[gene mutation]] is found in the majority of the patients with Wilson's disease. | |||
==Pathophysiology== | ==Pathophysiology== | ||
=== Normal copper transportation and metabolism === | === Normal copper transportation and metabolism === | ||
* [[Copper]] is one of the essential elements that is required daily in [[diet]] in a range of 1.5 to 2 mg.<ref name="pmid6280488">{{cite journal| author=Sandstead HH| title=Copper bioavailability and requirements. | journal=Am J Clin Nutr | year= 1982 | volume= 35 | issue= 4 | pages= 809-14 | pmid=6280488 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=6280488 }}</ref> | |||
* Copper is incorporated in number of important functions in the body. It functions as a component of some [[enzymes]] as [[cytochrome c oxidase]], [[dopamine beta hydroxylase|dopamine β-hydroxylase]], [[superoxide dismutase]] and [[tyrosinase]].<ref name="pmid62804882">{{cite journal| author=Sandstead HH| title=Copper bioavailability and requirements. | journal=Am J Clin Nutr | year= 1982 | volume= 35 | issue= 4 | pages= 809-14 | pmid=6280488 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=6280488 }}</ref> | |||
* In order to function appropriately, a significant proportion of [[copper]] must be absorbed and transported to the different site of actions. [[Stomach]] and [[duodenum]] are the main sites of absorption of copper then it binds [[albumin]] and transported to different [[Body tissue|body tissues]]. The excess copper is excreted as [[feces]] after being a part of the [[bile]]. | |||
* Copper is carried inside the [[cells]] by a transporter [[protein]] on the [[enterocyte|cells of the small bowel]] called [[SLC31A1|copper membrane transporter 1]] (CMT1). Others are bound to [[metallothionein]] where the rest is carried by [[ATOX1]] to the [[Golgi apparatus|trans-Golgi network]]. | |||
* Transportation of the [[copper]] to different [[Body tissue|body tissues]] is regulated by [[ATP7B|ATP7B gene]] in the [[hepatocytes]]. | |||
* [[ATP7B|ATP7B gene]] acts on transporting [[copper]] via two ways:<ref name="pmid17390257">{{cite journal| author=Pfeiffer RF| title=Wilson's Disease. | journal=Semin Neurol | year= 2007 | volume= 27 | issue= 2 | pages= 123-32 | pmid=17390257 | doi=10.1055/s-2007-971173 | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=17390257 }}</ref><ref name="pmid9261163">{{cite journal| author=Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD| title=Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. | journal=J Biol Chem | year= 1997 | volume= 272 | issue= 34 | pages= 21461-6 | pmid=9261163 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=9261163 }} </ref> | |||
** In the [[Trans Golgi network|trans-golgi network]]: Formation of [[ceruloplasmin]] (which is secreted into the [[blood]]) by adding the copper to the apoceruloplasmin. | |||
** In the [[cytoplasmic]] [[vesicles]]: Excretion of the excess copper into [[Bile|the bile]] by [[exocytosis]] against the [[hepatocytes]] memebrane. | |||
[[Image:Copper metabolism.png|thumb|center|framed|Normal absorption and distribution of copper. Cu = copper, CP = ceruloplasmin, green = ATP7B carrying copper. Source:By en:User:Jfdwolff - Copper Metabolism, English Wikipedia, Public Domain, https://commons.wikimedia.org/w/index.php?curid=7777267]] | |||
=== Pathogenesis === | === Pathogenesis === | ||
* The following table includes the main mechanisms that play an important role in pathogenesis of Wilson's disease. | |||
{| class="wikitable" | |||
!Impaired copper metabolism | |||
!Role in Wilson's disease pathogenesis | |||
|- | |||
|Impaired [[copper]] incorporation | |||
| | |||
* Failure of the copper to be incorporated into apoceruloplasmin leads to accumulation of the [[copper]] in the [[hepatocytes]] and different [[Body tissue|body tissues]] and [[organs]]. | |||
* [[Ceruloplasmin]] level may be decreased as a result of impaired copper incorporation. However, it is not required to diagnose Wilson's disease. | |||
|- | |||
|Copper accumulation in hepatocytes | |||
| | |||
* The copper accumulates in the [[hepatocytes]] and ends up into [[Liver cells|liver cell]] [[injury]]. | |||
|- | |||
|Extrahepatic copper accumulation | |||
| | |||
The | * Copper accumulation will eventually lead to copper deposition in other organs like [[Brain|the brain]]. | ||
* The [[urinary]] excretion of the [[copper]] will not be able to decrease the serum levels and the deposition increases over time ending by the damage of the other [[organs]] especially the [[brain]]. | |||
|} | |||
== | == Genetics == | ||
* [[ATP7B]] [[gene mutation]] is the main cause of [[copper]] transportation impairment and Wilson's disease. | |||
* ATP7B gene placed on [[chromosome 13]] and is expressed primarily in the [[liver]], [[kidney]], and [[placenta]]. | |||
* Wilson's disease [[ATP7B]] [[gene mutation]] is inhereted in an [[Autosomal recessive|autosomal recessive pattern]]. The majority of the patients have no family history. | |||
== Associated conditions == | == Associated conditions == | ||
* Wilson's disease may be associated with the following conditions:<ref name="de BieMuller2007">{{cite journal|last1=de Bie|first1=P|last2=Muller|first2=P|last3=Wijmenga|first3=C|last4=Klomp|first4=L W J|title=Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes|journal=Journal of Medical Genetics|volume=44|issue=11|year=2007|pages=673–688|issn=1468-6244|doi=10.1136/jmg.2007.052746}}</ref> | |||
** [[Fulminant liver failure]] | |||
** [[Liver cirrhosis|Cirrhosis]] | |||
** [[Aceruloplasminaemia]] | |||
==Gross pathology== | ==Gross pathology== | ||
| Line 59: | Line 70: | ||
</div> | </div> | ||
==Microscopic pathology== | ==Microscopic pathology== | ||
* Histological examination of a [[Liver biopsy|liver]] biopsy may show the following:<ref name="pmid167753002">{{cite journal| author=Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW et al.| title=MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. | journal=AJNR Am J Neuroradiol | year= 2006 | volume= 27 | issue= 6 | pages= 1373-8 | pmid=16775300 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=16775300 }}</ref> | |||
** Mild [[steatosis]] which is considered an early histological feature | |||
** Glycogenated [[hepatic]] [[nuclei]] | |||
** [[Hepatocellular Disease|Hepatocellular]] necrosis | |||
** Autoimmune hepatitis histologic features | |||
** Fibrosis and cirrhosis (macronodular or micronodular) in advanced cases | |||
** Fulminant liver falilure features which include: | |||
*** Hepatocellular degenration | |||
*** Parenchymal collapse | |||
==References== | ==References== | ||
| Line 67: | Line 87: | ||
{{WH}} | {{WH}} | ||
{{WS}} | {{WS}} | ||
[[Category:Gastroenterology]] | |||
[[Category:Hematology]] | |||
[[Category:Medicine]] | |||
[[Category:Up-To-Date]] | |||
Latest revision as of 19:18, 29 August 2018
| https://https://www.youtube.com/watch?v=Cr8R_bnKAtk%7C350}} |
|
Wilson's disease Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Wilson's disease pathophysiology On the Web |
|
American Roentgen Ray Society Images of Wilson's disease pathophysiology |
|
Risk calculators and risk factors for Wilson's disease pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Copper is one of the essential elements for the human body. Copper must be absorbed and transported in order to function properly. Copper is transported bound to metallothionein or carried by ATOX1 to the trans-Golgi network. Impairment of copper incorportation in formation of ceruloplasmin will lead to increase the serum level of the copper. The increase of copper level will lead to accumulation of the excess amount in the liver and other organs. ATP7B gene mutation is found in the majority of the patients with Wilson's disease.
Pathophysiology
Normal copper transportation and metabolism
- Copper is one of the essential elements that is required daily in diet in a range of 1.5 to 2 mg.[1]
- Copper is incorporated in number of important functions in the body. It functions as a component of some enzymes as cytochrome c oxidase, dopamine β-hydroxylase, superoxide dismutase and tyrosinase.[2]
- In order to function appropriately, a significant proportion of copper must be absorbed and transported to the different site of actions. Stomach and duodenum are the main sites of absorption of copper then it binds albumin and transported to different body tissues. The excess copper is excreted as feces after being a part of the bile.
- Copper is carried inside the cells by a transporter protein on the cells of the small bowel called copper membrane transporter 1 (CMT1). Others are bound to metallothionein where the rest is carried by ATOX1 to the trans-Golgi network.
- Transportation of the copper to different body tissues is regulated by ATP7B gene in the hepatocytes.
- ATP7B gene acts on transporting copper via two ways:[3][4]
- In the trans-golgi network: Formation of ceruloplasmin (which is secreted into the blood) by adding the copper to the apoceruloplasmin.
- In the cytoplasmic vesicles: Excretion of the excess copper into the bile by exocytosis against the hepatocytes memebrane.

Pathogenesis
- The following table includes the main mechanisms that play an important role in pathogenesis of Wilson's disease.
| Impaired copper metabolism | Role in Wilson's disease pathogenesis |
|---|---|
| Impaired copper incorporation |
|
| Copper accumulation in hepatocytes |
|
| Extrahepatic copper accumulation |
Genetics
- ATP7B gene mutation is the main cause of copper transportation impairment and Wilson's disease.
- ATP7B gene placed on chromosome 13 and is expressed primarily in the liver, kidney, and placenta.
- Wilson's disease ATP7B gene mutation is inhereted in an autosomal recessive pattern. The majority of the patients have no family history.
Associated conditions
- Wilson's disease may be associated with the following conditions:[5]
Gross pathology
-
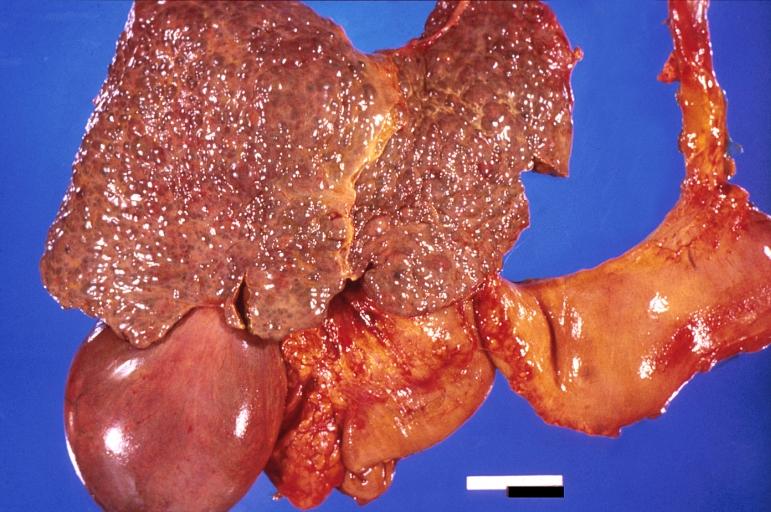
Liver cirrhosis and enlarged gall bladder in Wilson's disease.
-
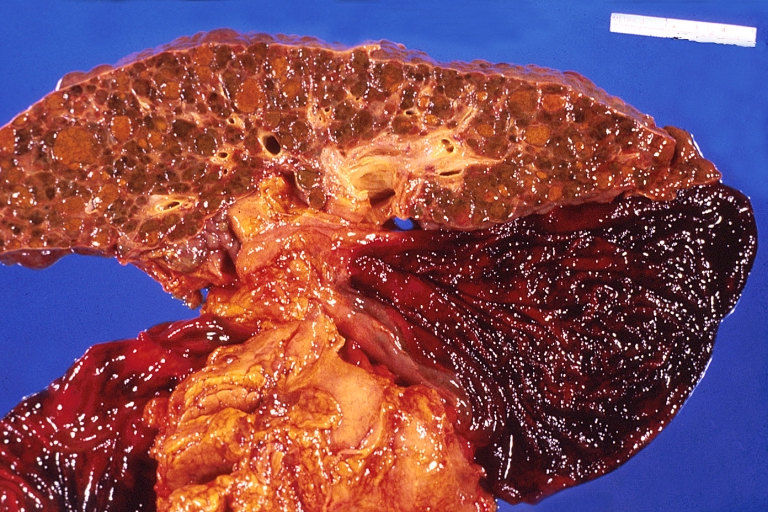
Liver cirrhosis and enlarged gall bladder in Wilson's disease.
-
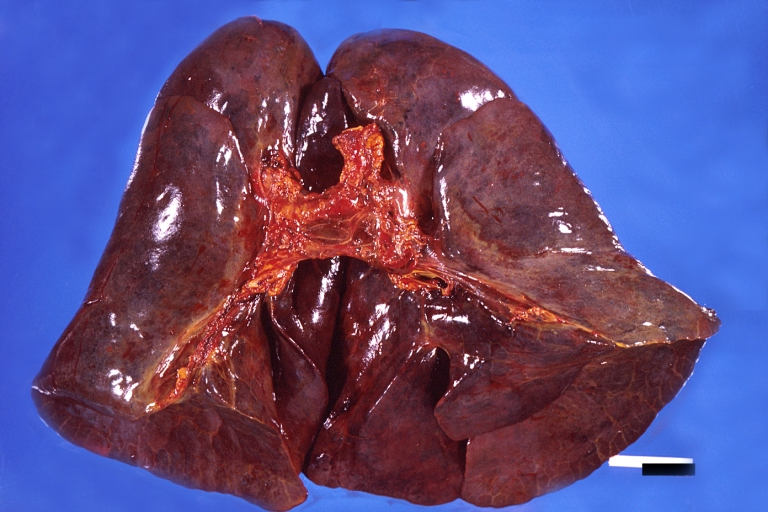
Lung, hemorrhagic bronchopneumonia, Wilson's disease
-
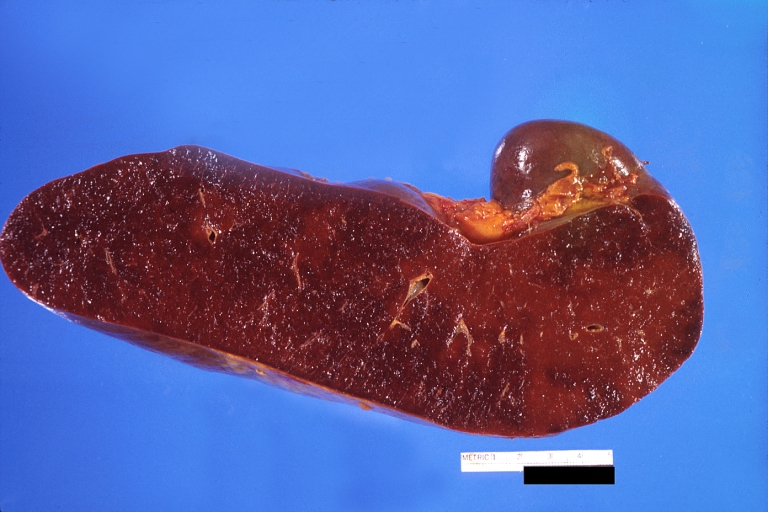
Spleen, congestion. Wilson's disease




Microscopic pathology
- Histological examination of a liver biopsy may show the following:[6]
- Mild steatosis which is considered an early histological feature
- Glycogenated hepatic nuclei
- Hepatocellular necrosis
- Autoimmune hepatitis histologic features
- Fibrosis and cirrhosis (macronodular or micronodular) in advanced cases
- Fulminant liver falilure features which include:
- Hepatocellular degenration
- Parenchymal collapse
References
- ↑ Sandstead HH (1982). "Copper bioavailability and requirements". Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Sandstead HH (1982). "Copper bioavailability and requirements". Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Pfeiffer RF (2007). "Wilson's Disease". Semin Neurol. 27 (2): 123–32. doi:10.1055/s-2007-971173. PMID 17390257.
- ↑ Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997). "Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae". J Biol Chem. 272 (34): 21461–6. PMID 9261163.
- ↑ de Bie, P; Muller, P; Wijmenga, C; Klomp, L W J (2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". Journal of Medical Genetics. 44 (11): 673–688. doi:10.1136/jmg.2007.052746. ISSN 1468-6244.
- ↑ Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW; et al. (2006). "MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation". AJNR Am J Neuroradiol. 27 (6): 1373–8. PMID 16775300.