Tuberculosis pathophysiology
|
Tuberculosis Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Tuberculosis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Tuberculosis pathophysiology |
|
Risk calculators and risk factors for Tuberculosis pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: João André Alves Silva, M.D. [2]
Overview
M. tuberculosis is acquired by inhaled aerosols produced by patients with active disease. The mycobacterium thrives in the upper lung lobes given the high oxygen content. Tuberculosis is a prototypical granulomatous infection. The granuloma prevents the dissemination of mycobacteria and provides a pathway for immune cell communication. Within the granuloma, CD4 T lymphocytes secrete cytokines such as interferon gamma that activate local macrophages.
Pathogenesis
The M. tuberculosis bacterium is acquired by inhaled aerosols generated by individuals with active pulmonary disease. They travel to the terminal airways and alveoli (commonly at the middle lobes, upper regions of lower lobes and lower regions of upper lobes) and are phagocytosed by alveolar macrophages. The initial immune response generated by these macrophages recruits further macrophages, neutrophils, and monocytes, leading to primary containment of the bacilli. Despite having a very low infectious dose (ID<200 bacteria), 90% of immunocompetent individuals that acquire M. tuberculosis do not develop symptoms. In most cases, the bacteria may either be eliminated, or be harbored in a latent state by an immune formation known as a granuloma. The granuloma is a structured, radial aggregation of immune cells that prevents the dissemination of mycobacteria and provides a pathway for immune cell communication.[1] The initial focus of infection in the lung is a single region in 75% of the cases, and is called the Ghon focus. Besides the alveolar macrophages, other immune cells such as blood monocytes (tissue macrophages) and lymphocytes also migrate to the Ghon focus aiding in the termination of the infection or initiation of a granulomatous containment.[1][2]
Primary Infection
In an immunocompetent person, the infected macrophages are transported through the lymph to the regional lymph nodes. In immunocompromised patients, these macrophages may reach different parts of the body, through the bloodstream. Despite this wide initial dissemination, most patients resolve those foci of infection without any signs or symptoms of disease. However, during the dissemination of the infected macrophages, tissues that are more prone to bacterial replication, represent potential metastatic foci. These tissues include:[1][2]
- Apical-posterior regions of the lungs
- Lymph nodes
- Kidneys
- Vertebral bodies
- Extremities of long bones
- Juxta ependymal meningeal regions
Although all parts of the body can be affected by TB, the heart, skeletal muscles, pancreas and thyroid are rarely affected.[3] In a small number of cases, when there is a large concentration of antigens in the primary focus, the development of the immune response and hypersensitivity may lead to the necrosis and calcification of this infection site. These primary calcified foci are called Ranke complex.[1][4]
Progression of the Primary Infection
Initial foci of infection may evolve into large pulmonary lymph nodes. These may lead to:[1]
- Bronchial collapse
- Formation of atelectasis
- Erosion of the bronchus, with further spread of infection
More commonly in non-caucasian children, with inferior resistance to tuberculosis, the primary focus of infection may evolve to constitute progressive primary disease, with advancing pneumonia. The infection may lead to the formation of cavitations with spread of the infection through the bronchi. This may also occur in HIV and elderly patients.[1][5][6] In young children, the dissemination of infection before the onset of hypersensitivity may lead to military tuberculosis. Bacteria may disseminate directly from the primary focus, or from the Weigart focus (metastatic focus adjacent to a pulmonary vein) through the blood.[1][7] In younger patients, serofibrinous pleurisy is more prone to occur following the rupture of subpleural foci of infection into the pleural space.[1] The most severe consequence of the dissemination of bacteria from primary or metastatic foci, through the blood and lymph, is the seeding of the postero-apical regions of the lung. Here bacteria are able to replicate without the opposition of the immune system, potentially leading pulmonary tuberculosis.[1]
Immune Response
The immune response against M. tuberculosis has a great involvement from cellular immunity.[8] Despite the humural response during tuberculosis with production of numerous antibodies, its role in the immune response towards the bacteria is not defined. The immune response against M. tuberculosis is minimal during the first weeks, allowing it to replicate in the alveolar spaces and macrophages, constituting the Ghon focus, or metastatic foci. Bacterial entrance intro the macrophages occurs by interaction with the following receptors:[9]
- Fc receptors
- Mannose receptors
- Complement receptors
Molecular Pathogenesis
- Interferon gamma and tumor necrosis factor alpha signaling activates the macro-phages which engulf mycobacterium tuberculosis. [10]
- Metalloproteinase converts the trans membrane protein to soluble tumor necrosis factor. This binds with the receptor TNFR1 and TNFR2 and through caspase dependant pathways induce apoptosis.
- TNF along with the synergistic action of interferon gamma increases the phagocytic activity of the macrophages and causes the intracellular killing of the pathogens by reactive nitrogen and oxygen intermediates.
- Neutralization of the TNF activity causes the persistence of mycobacterial activities within the granuloma in latent infection.Thus they are required for the formation and maintanence of granuloma in tuberculosis.
- TNF stimulates the production of CCL2, CCL3, CCL4, CCL5, CCL8 cytokines and increases CD54 causing focussed accumulation of immune cells and plays a pivotal role in the generation of granuloma and maintaining the integrity of the granuloma. [10]

Once within alveolar macrophages, M. tuberculosis uses multiple mechanisms in order to survive:[1]
- Urease - prevents acidification of macrophageal lysosomes, limiting action of cellular enzymes
- Secretion of antioxidants, for suppression of reactive oxygen species, such as:
Alveolar macrophages and dendritic cells present mycobacterial antigens on their surfaces through class II major histocompatibility complex. These antigens will be recognized by CD4 lymphocytes through αβ T-cell receptors. CD4 lymphocytes, once activated, release lymphokines that attract more macrophages to the site of infection. During lymphocyte activation, immune cells produce large amounts of lytic enzymes, which when released, lead to the necrosis of tissues. Macrophages will also stimulate epithelioid cells that will be responsible for the formation of the granuloma.[1]
About 3-9 weeks after the infection with M. tuberculosis, the immune response reaches a level that is manifested by a positive TST result. The concentration of mycobacterial antigens and the level of the immune response are responsible for the clinical manifestations of TB.[1]
A granuloma is formed in the presence of a low antigen load, with an hypersensitive tissue response, that is manifested by a large accumulation of host cells, such as:[1]
In the presence of a high antigen load, with a hypersensitive tissue response, immune cells are not properly organized, and necrotic tissue may be formed.[1]
When the reaction of the immune system is successful, the healing process leads to the formation of a scar on the fibrotic and encapsulated tissue (exudative reaction). If necrosis is incomplete, a caseous material may be formed.[12]
If the caseous material is discharged through the bronchial airways, tuberculous cavities may be formed and coinfected by multiple organisms. In the absence of necrosis, full healing may occur.[12]
If the immune response is weak, nonreactive TB may be noted. In this case, unspecific tissue changes may be noted, with few immune cells surrounding large amounts of bacilli.[13]
In persons with a positive TST, endogenous foci of bacteria may be reactivated. CD4 lymphocytes are responsible for inhibiting this reactivation.[14]
Transmission
After contact with a patient that has the active form of the disease, and inhalation of the M. tuberculosis the risk of developing active tuberculosis is low, with a life-time projected risk of about 10%.[15] The probability of transmission from one person to another depends on the number of infectious droplets expelled by the carrier, the effectiveness of ventilation, the duration of the exposure, and the virulence of the strain of M. tuberculosis.[16] The probability of transmitting the disease is highest during the first years, after the person has been infected, decreasing hence forth.[17]
In rare occasions, the bacteria may be transmitted through other routes, besides the pulmonary. In these cases, the formation of foci in local lymph nodes is always involved. These alternative routes of transmission include:[1]
- Skin abrasions
- Oropharynx
- Intestine
- Genitalia
Associated Conditions
AIDS
Tuberculosis influences the progression of HIV replication in infected patients, leading to an increase in the mortality rate.[18]
On the other hand, HIV infected patients, particularly those with low counts of CD4 lymphocytes, have increased risk of reactivation of latent tuberculosis. Additionally, when recently infected with M. tuberculosis, these patients tend to rapidly progress into active disease.[1][19][20] It is still not known if AIDS influences the risk of infection, when in contact with the M. tuberculosis.[1]
Patients with AIDS have increased risk of developing pulmonary and extrapulmonary tuberculosis. Extrapulmonary disease in this group of patients has characteristic manifestations, such as:[1]
- Higher frequency of disseminated disease[21]
- Rapid progression of the disease with widespread pulmonary infiltrates
- DIC and acute respiratory failure
- When present, tuberculous pleuritis occurs bilaterally
- Abdominal and mediastinal lymphadenopathy are common
- Increased risk of Immune reconstitution inflammatory syndrome (IRIS)[22]
- Common abscesses of:[1]
Gallery
-
![Left lateral margin of a tongue of a tuberculosis patient, which had been retracted in order to reveal the lesion that had been caused by the Gram-positive bacterium Mycobacterium tuberculosisAdapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/0/09/TB1.jpg)
Left lateral margin of a tongue of a tuberculosis patient, which had been retracted in order to reveal the lesion that had been caused by the Gram-positive bacterium Mycobacterium tuberculosisAdapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection.[ http://phil.cdc.gov/phil/ Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.][23]](/images/e/ed/TB2.jpg)
Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection.[ http://phil.cdc.gov/phil/ Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.][23]
-
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/7/79/Leprosy-35.jpg)
Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/6/60/Leprosy-36.jpg)
Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/9/93/Leprosy-37.jpg)
Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/b/b5/Leprosy-38.jpg)
Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Photomicrograph describing tuberculosis of the placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/b/be/TB3.jpg)
Photomicrograph describing tuberculosis of the placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Histopathology of tuberculosis, endometrium. Ziehl-Neelsen stain.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/d/dd/TB4.jpg)
Histopathology of tuberculosis, endometrium. Ziehl-Neelsen stain.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
![Histopathology of tuberculosis, placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/images/9/97/TB5.jpg)
Histopathology of tuberculosis, placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]
-
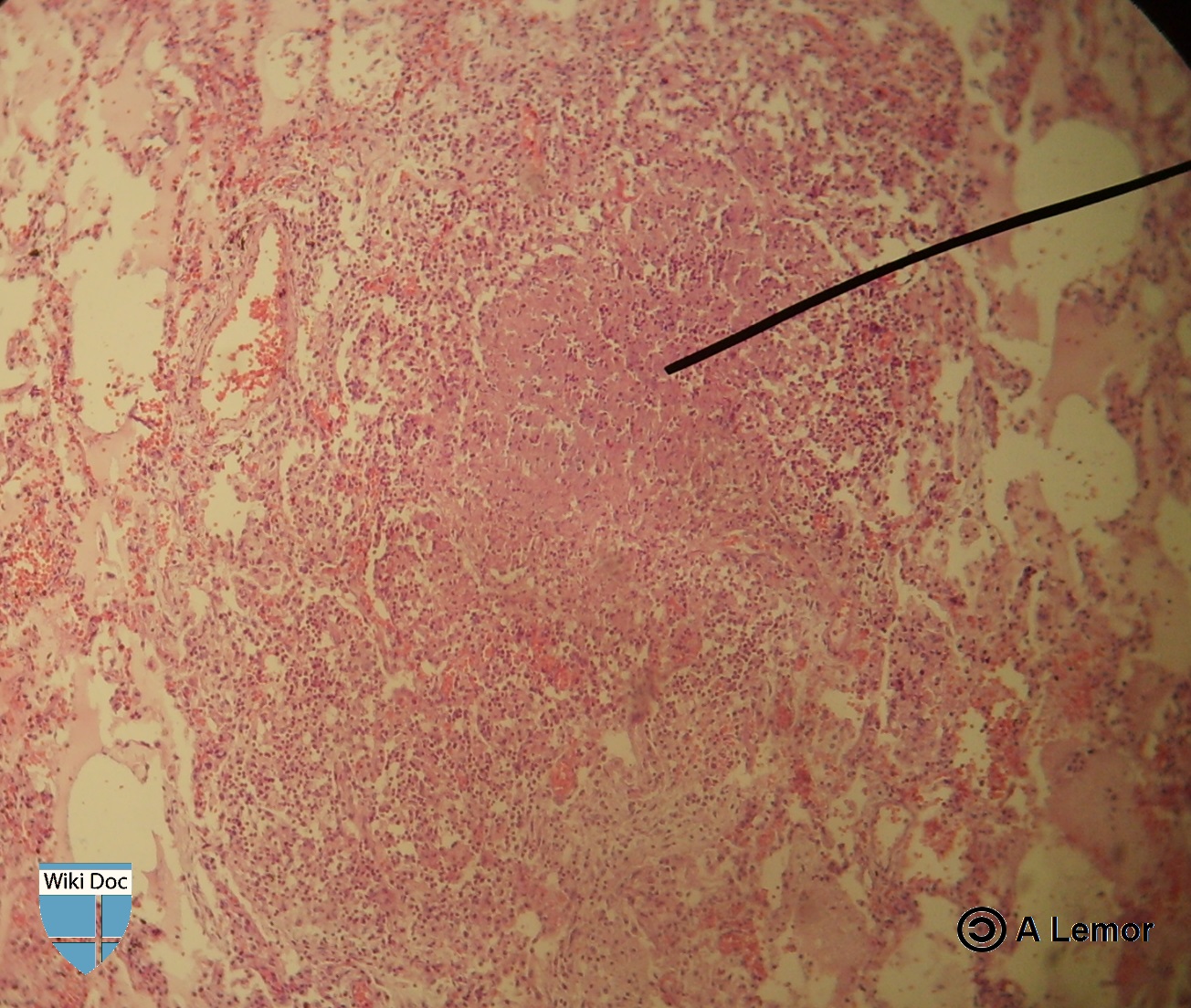
Miliar Tuberculosis
-
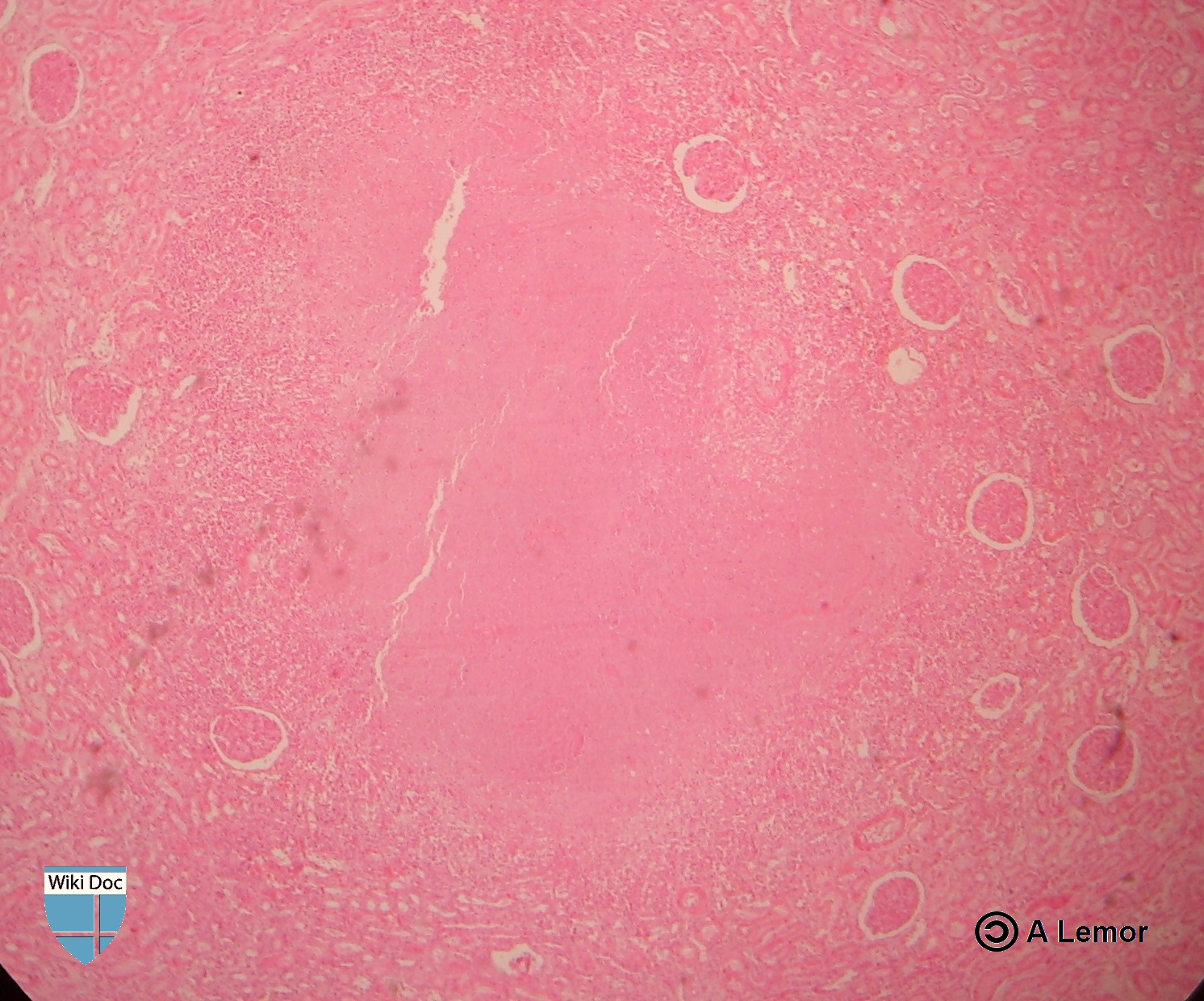
Renal Tuberculosis lesion
![Left lateral margin of a tongue of a tuberculosis patient, which had been retracted in order to reveal the lesion that had been caused by the Gram-positive bacterium Mycobacterium tuberculosisAdapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:TB1.jpg)
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection.[ http://phil.cdc.gov/phil/ Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.][23]](/index.php/File:TB2.jpg)
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:Leprosy-35.jpg)
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:Leprosy-36.jpg)
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:Leprosy-37.jpg)
![Light photomicrograph revealing some of the histopathologic cytoarchitectural characteristics seen in a mycobacterial skin infection Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:Leprosy-38.jpg)
![Photomicrograph describing tuberculosis of the placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:TB3.jpg)
![Histopathology of tuberculosis, endometrium. Ziehl-Neelsen stain.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:TB4.jpg)
![Histopathology of tuberculosis, placenta.Adapted from Public Health Image Library (PHIL), Centers for Disease Control and Prevention.[23]](/index.php/File:TB5.jpg)


{kind=link}
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 Mandell, Gerald (2010). Mandell, Douglas, and Bennett's principles and practice of infectious diseases. Philadelphia, PA: Churchill Livingstone/Elsevier. ISBN 0443068399.
- ↑ 2.0 2.1 Herrmann J, Lagrange P (2005). "Dendritic cells and Mycobacterium tuberculosis: which is the Trojan horse?". Pathol Biol (Paris). 53 (1): 35–40. PMID 15620608.
- ↑ Agarwal R, Malhotra P, Awasthi A, Kakkar N, Gupta D (2005). "Tuberculous dilated cardiomyopathy: an under-recognized entity?". BMC Infect Dis. 5 (1): 29. PMID 15857515.
- ↑ Grosset J (2003). "Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary". Antimicrob Agents Chemother. 47 (3): 833–6. PMID 12604509.
- ↑ Stead WW, Lofgren JP, Warren E, Thomas C (1985). "Tuberculosis as an endemic and nosocomial infection among the elderly in nursing homes". N Engl J Med. 312 (23): 1483–7. doi:10.1056/NEJM198506063122304. PMID 3990748.
- ↑ Murray JF (1990). "Cursed duet: HIV infection and tuberculosis". Respiration. 57 (3): 210–20. PMID 2274719.
- ↑ Kim J, Park Y, Kim Y, Kang S, Shin J, Park I, Choi B (2003). "Miliary tuberculosis and acute respiratory distress syndrome". Int J Tuberc Lung Dis. 7 (4): 359–64. PMID 12733492.
- ↑ Orme IM, Andersen P, Boom WH (1993). "T cell response to Mycobacterium tuberculosis". J Infect Dis. 167 (6): 1481–97. PMID 8501346.
- ↑ Aderem A, Underhill DM (1999). "Mechanisms of phagocytosis in macrophages". Annu Rev Immunol. 17: 593–623. doi:10.1146/annurev.immunol.17.1.593. PMID 10358769.
- ↑ 10.0 10.1 "Tumor Necrosis Factor alpha".
- ↑ "TNF Alpha". Missing or empty
|url=(help) - ↑ 12.0 12.1 Canetti G (1965). "Present aspects of bacterial resistance in tuberculosis". Am Rev Respir Dis. 92 (5): 687–703. PMID 5321145.
- ↑ O'BRIEN JR (1954). "Non-reactive tuberculosis". J Clin Pathol. 7 (3): 216–25. PMC 1023795. PMID 13192197.
- ↑ Ellner JJ (1997). "Review: the immune response in human tuberculosis--implications for tuberculosis control". J Infect Dis. 176 (5): 1351–9. PMID 9359738.
- ↑ Glaziou P, Falzon D, Floyd K, Raviglione M (2013). "Global epidemiology of tuberculosis". Semin Respir Crit Care Med. 34 (1): 3–16. doi:10.1055/s-0032-1333467. PMID 23460002.
- ↑ "Causes of Tuberculosis". Mayo Clinic. 2006-12-21. Retrieved 2007-10-19.
- ↑ Lawn SD, Zumla AI (2011). "Tuberculosis". Lancet. 378 (9785): 57–72. doi:10.1016/S0140-6736(10)62173-3. PMID 21420161.
- ↑ Zumla A, Raviglione M, Hafner R, von Reyn CF (2013). "Tuberculosis". N Engl J Med. 368 (8): 745–55. doi:10.1056/NEJMra1200894. PMID 23425167.
- ↑ Daley CL, Small PM, Schecter GF, Schoolnik GK, McAdam RA, Jacobs WR; et al. (1992). "An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction-fragment-length polymorphisms". N Engl J Med. 326 (4): 231–5. doi:10.1056/NEJM199201233260404. PMID 1345800.
- ↑ Bouvet E, Casalino E, Mendoza-Sassi G, Lariven S, Vallée E, Pernet M; et al. (1993). "A nosocomial outbreak of multidrug-resistant Mycobacterium bovis among HIV-infected patients. A case-control study". AIDS. 7 (11): 1453–60. PMID 8280411.
- ↑ Shafer RW, Kim DS, Weiss JP, Quale JM (1991). "Extrapulmonary tuberculosis in patients with human immunodeficiency virus infection". Medicine (Baltimore). 70 (6): 384–97. PMID 1956280.
- ↑ Meintjes G, Lawn SD, Scano F, Maartens G, French MA, Worodria W; et al. (2008). "Tuberculosis-associated immune reconstitution inflammatory syndrome: case definitions for use in resource-limited settings". Lancet Infect Dis. 8 (8): 516–23. doi:10.1016/S1473-3099(08)70184-1. PMC 2804035. PMID 18652998.
- ↑ 23.0 23.1 23.2 23.3 23.4 23.5 23.6 23.7 23.8 "Public Health Image Library (PHIL), Centers for Disease Control and Prevention".