Pheochromocytoma pathophysiology
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma pathophysiology |
|
Risk calculators and risk factors for Pheochromocytoma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2] Mohammed Abdelwahed M.D[3]
Overview
Pheochromocytoma arises from chromaffin cells of the adrenal medulla.On gross pathology, pheochromocytoma has a multinodular and a multicentric pattern of growth. On microscopic histopathological analysis, nesting (Zellballen) pattern is composed of well-defined clusters of tumor cells separated by fibrovascular stroma. It may be benign, malignant, familial(multiple endocrine neoplasia 1 and type 2B) or sporadic. All of these forms have genetic origin depending on a large number of genes, for example, VHL, SDH, NF1, RET genes.
Pathophysiology
- Pheochromocytoma arises from chromaffin cells of the adrenal medulla and sympathetic ganglia. Malignant and benign pheochromocytomas share the same biochemical and histological features, the only difference is to have a distant spread or be locally invasive. [1]
Basic physiology of catecholamines
- Epinephrine acts on nearly all body tissues. Its actions vary by tissue type and tissue expression of adrenergic receptors.
- Epinephrine is a nonselective agonist of all adrenergic receptors, including the major subtypes α1, α2, β1, β2, and β3:
- Binding to α1 receptors causes vasoconstriction. Blood vessels with α1-adrenergic receptors are present in the skin, the sphincters of the gastrointestinal system, kidney (renal artery) and brain. During the fight-or-flight response vasoconstriction results in decreased blood flow to these organs.
- Binding to α2 receptors inhibits insulin secretion by the pancreas, stimulates glycogenolysis in the liver and muscle, and stimulates glycolysis and inhibits insulin-mediated glycogenesis in muscle. It suppresses the release of norepinephrine by negative feedback.
- Binding to B2 receptors causes Smooth muscle relaxation in the uterus, GI tract, detrusor urinae muscle of bladder wall, and bronchi. It also causes dilatation of smaller coronary arteries, hepatic artery, arteries to skeletal muscle.
- Binding to B1 receptors causes renin release from juxtaglomerular cells and lipolysis in adipose tissue. It Increases cardiac output by:
- Increase in heart rate in sinoatrial node
- Increase in atrial cardiac muscle contractility
- Increases in contractility and automaticity of ventricular cardiac muscle
- Increases in conduction and automaticity of atrioventricular node
Pathogenesis
Genetics
- Pheochromocytomas can be familial and occur in patients with multiple endocrine neoplasias (MEN1 and MEN 2B).
- Patients with Von Hippel Lindau disease (VHL) may also develop pheochromocytoma.[4]
- It has autosomal dominant inheritance and has two pathways of tumor pathogenesis. Cluster 1 tumors are noradrenergic. Cluster 2 tumors are adrenergic.[5]
| Familial pheocromocytomas | |
|---|---|
| Cluster 1 (Noradrenergic) | Cluster 2 (Adrenergic) |
|
|
- Patients with the succinate dehydrogenase B mutations are likely to develop a malignant disease.[6]
Associated conditions
- Pheochromocytoma can be part of other syndromes named multiple endocrine neoplasias (MEN1 and MEN2B), which are autosomal dominant syndromes controlled by RET gene. Pheochromocytoma occurs in 50% of patients with MEN2 as follows:
| MEN1 | MEN2 |
|---|---|
Gross Pathology
On gross pathology, A multinodular and multicentric pattern of growth of pheochromocytoma may be seen.
-
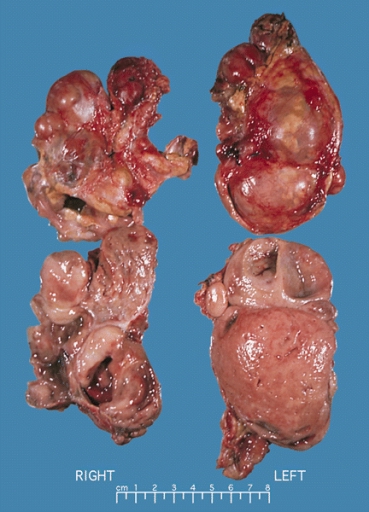
Bilateral pheochromocytoma in MEN2. Gross image.

Microscopic Pathology
On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma.
-
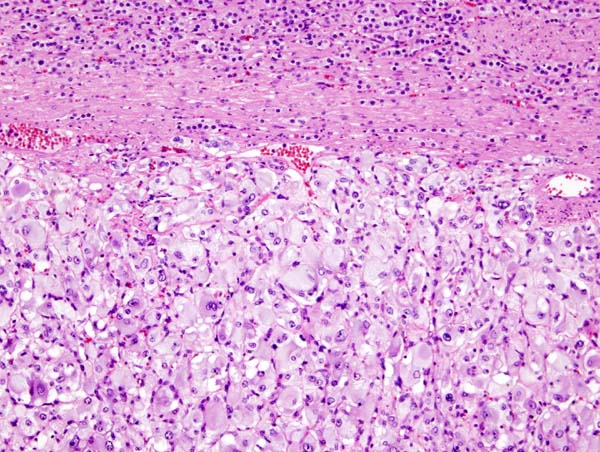
Micrograph of pheochromocytoma.
-
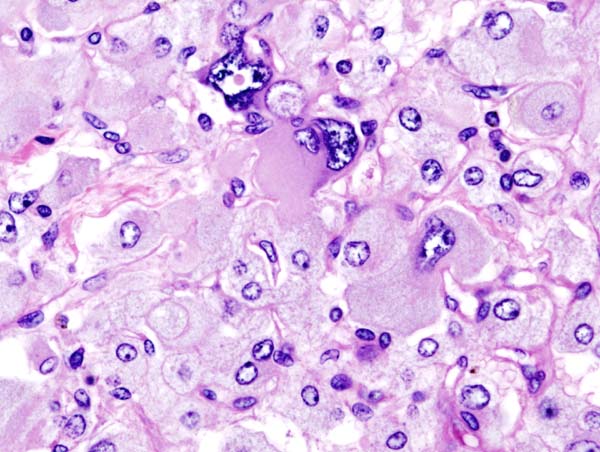
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen.
-
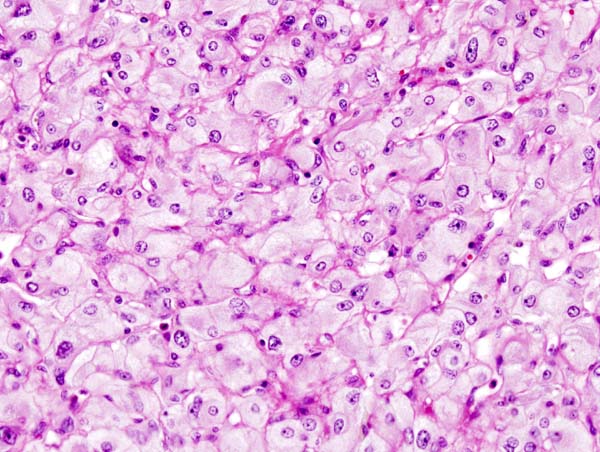
Micrograph of pheochromocytoma.
-
Micrograph of pheochromocytoma.
_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
Videos
{{#ev:youtube|7yjxG3KmX98}}
References
- ↑ Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA; et al. (1999). "Clinical experience over 48 years with pheochromocytoma". Ann Surg. 229 (6): 755–64, discussion 764-6. PMC 1420821. PMID 10363888.
- ↑ Webb TA, Sheps SG, Carney JA (1980). "Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2". Am. J. Surg. Pathol. 4 (2): 121–6. PMID 6103678.
- ↑ Yee JK, Moores JC, Jolly DJ, Wolff JA, Respess JG, Friedmann T (1987). "Gene expression from transcriptionally disabled retroviral vectors". Proc. Natl. Acad. Sci. U.S.A. 84 (15): 5197–201. PMC 298821. PMID 3474647.
- ↑ Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM (2014). "The genetic basis of pheochromocytoma and paraganglioma: implications for management". Urology. 83 (6): 1225–32. doi:10.1016/j.urology.2014.01.007. PMC 4572836. PMID 24642075.
- ↑ King KS, Pacak K (2014). "Familial pheochromocytomas and paragangliomas". Mol Cell Endocrinol. 386 (1–2): 92–100. doi:10.1016/j.mce.2013.07.032. PMC 3917973. PMID 23933153.
- ↑ Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M; et al. (2004). "Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations". JAMA. 292 (8): 943–51. doi:10.1001/jama.292.8.943. PMID 15328326.