Timothy syndrome
|
WikiDoc Resources for Timothy syndrome |
|
Articles |
|---|
|
Most recent articles on Timothy syndrome Most cited articles on Timothy syndrome |
|
Media |
|
Powerpoint slides on Timothy syndrome |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Timothy syndrome at Clinical Trials.gov Trial results on Timothy syndrome Clinical Trials on Timothy syndrome at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Timothy syndrome NICE Guidance on Timothy syndrome
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Timothy syndrome Discussion groups on Timothy syndrome Patient Handouts on Timothy syndrome Directions to Hospitals Treating Timothy syndrome Risk calculators and risk factors for Timothy syndrome
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Timothy syndrome |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Vamsikrishna Gunnam M.B.B.S [2]
Synonyms and keywords: Long QT syndrome 8; LQT8; Long QT syndrome with syndactyly; TS
Overview
Timothy syndrome is a rare syndrome that follows autosomal dominant inheritance pattern. Timothy syndrome is a multisystem disorder characterized by physiological and developmental defects which include long QT-prolongation, arrhythmias, structural heart defects, syndactyly and autism spectrum disorders. Timothy syndrome may be classified into 2 groups, classical form(type-1) and atypical form(type-2). Timothy syndrome caused by mutations in CACNA1C, which encodes for calcium channel α subunit. Timothy syndrome often ends in early death. The United States of America in order to categorize a condition as a rare disease it should affect fewer than 200,000 people. Rare diseases also called as orphan diseases. Orphan Drug Act was passed in 1983 by congress for the rare diseases. Today an average of 25-30 million Americans have been reported with rare diseases. The number of people with individual rare disease may be less but overall the number of people with rare diseases are large in number.
Historical Perspective
- In 1995, Splawski, Reichenbach, and Marks were the first to give the name Timothy syndrome in the honor of Dr.Katherine W. Timothy who did the phenotypic analysis.[3]
Classification
| Type | Gene | Protein involved | Location | Mutations | Inheritance pattern | Symptoms |
|---|---|---|---|---|---|---|
| Classic Timothy syndrome | CACNA1C | Voltage-dependent L-type calcium channel subunit alpha-1C | 12p13.33 | Exon 8 a | Autosomal dominant | Long QT syndrome with syndactyly |
| Atypical Timothy syndrome | CACNA1C | Voltage-dependent L-type calcium channel subunit alpha-1C | 12p13.33 | Exon 8 | Autosomal dominant | A very severe form of long QT syndrome without syndactyly |
Pathophysiology
- It is understood that Timothy syndrome both classical and atypical form is the result of a missense mutation in the CACNA1C gene.[8][9][10]
- CACNA1C gene encodes for calcium channel α subunit across cell membranes.
- Any missense mutation in exon 8 (atypical form) and exon 8a (classical form) of CACNA1C gene results in structural changes of Ca(V)1.2channels and delayed calcium channel α subunit closing and, thus, increased cellular excitability.[11][12][13]
- There is one more mutation in G406R that is associated with the timothy syndrome.[14][15]
- The location of G406R is in domain one segment six (D1S6) which holds glycine at this position and plays a very important role in voltage-dependent inactivation.
- Calcium channel α subunit especially Ca(V)1.2 involved in transporting positively charged calcium ions into the cells across a cell membrane which plays a critical role in the normal function of heart and brain cells.
- Mutations in Ca(V)1.2 calcium channels leads to disruption of the following events in the heart and other organs:[16]
- Cell-to-cell communication
- Muscle contraction
- Normal regulation of certain genes
- Due to the fact that exon 8(atypical Timothy syndrome) is more expressed in heart muscles than that of exon 8a(classic Timothy syndrome) patients with exon 8 mutation have a severe form of long QT interval.
Causes
Genetic Causes
Differentiating Timothy syndrome from other Diseases
- Timothy syndrome must be differentiated from Jervell and Lange-Nielsen syndrome (JLNS), Romano-Ward syndrome, , Andersen-Tawil syndrome, Brugada syndrome, and Sudden infant death syndrome (SIDS).[19][20][21][22][23][24][25]
Epidemiology and Demographics
Incidence
- The incidence of timothy syndrome is unknown worldwide.
- Only 20 cases were reported worldwide.
Prevalence
- The prevalence of timothy syndrome is less than 1 per 100,000 individuals worldwide.
Mortality rate
- In patients with timothy syndrome the average age of death is 2.5 years
Age
- Timothy syndrome commonly affects individuals of younger age group, the median age of diagnosis is usally within the first few days after birth.
Race
- There is no racial predilection to Timothy syndrome.
Gender
- Timothy syndrome affects men and women equally.
Risk Factors
- There are no established risk factors for Timothy syndrome.
Screening
- There is insufficient evidence to recommend routine screening for Timothy syndrome.
Natural History, Complications, Prognosis
Natural History
- The symptoms of Timothy syndrome usually develop in the first decade of life, and start with symptoms such as cardiac, hand/foot, facial, and neurodevelopmental symptoms.
Complications
- Common complications of Timothy syndrome include:[26]
- Ventricular tachyarrhythmia which includes both ventricular tachycardia and ventricular fibrillation is the cause of death in most of the patients with Timothy syndrome
- Severe infections due to weakened immune system(despite aggressive antibiotic therapy)
- Intractable hypoglycemia[27]
Prognosis
- The prognosis for patients diagnosed with Timothy syndrome is grim.
Diagnosis
Diagnostic study of choice
- Sequence analysis(100% detectable), Targeted analysis for pathogenic variants(>95% detectable), Gene-targeted deletion/duplication analysis of CACNA1C gene testing is the gold standard test for the diagnosis of Timothy syndrome.[30]
- Along with the genetic testing patients also evaluate for following recommendations:
- Electrocardiogram(ECG)
- Echocardiogram
- Developmental and neurological assessment for autism and Intellectual disability
- Skeletal abnormalities
Symptoms
Common Symptoms
Common symptoms of Timothy syndrome include:[31][32]
- Torsade de pointes
- Syncopal events usually self-terminating
- Arrhythmias
- Palpitations
- Neuropsychiatric symptoms such as autism and Intellectual disability
- Seizures
- Hypotonia
Less Common Symptoms
Less common symptoms of Timothy syndrome include:[33][34]
Electrocardiogram
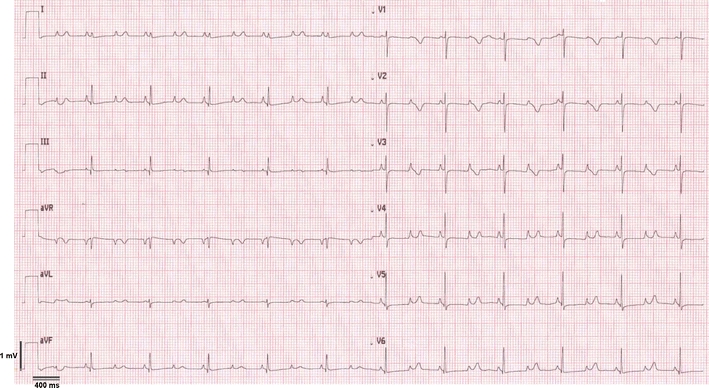
Trimonth syndrome ECG after birth showed a prolonged QT interval (QTc 600 ms), 2:1 atrioventricular block and significant bradycardia (ventricular rate 60/min). Case courtesy by U. Krause Et Al[35] 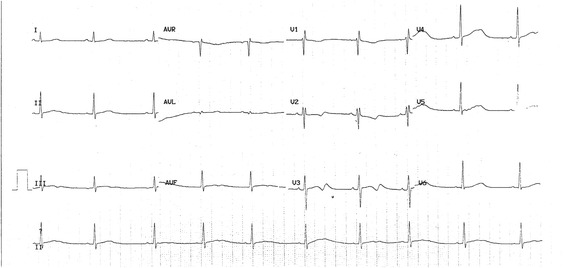
A boy with a heart rate at 58/min, QT 498 ms and QTc 490 ms in long Qt syndrome. Case courtesy by Thomas Hof Et Al[39] - Prolongation of the QTc interval with an average QTc of 640 ms in atypical Timothy syndrome
- Macroscopic T-wave alternans
- Tachyarrhythmias: due to abnormal cardiac depolarization and cardiac repolarization
- Ventricular tachycardia
- AV block
- Torsade de pointes ventricular tachycardia[40]
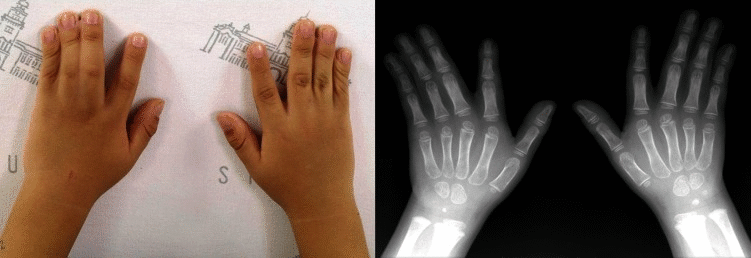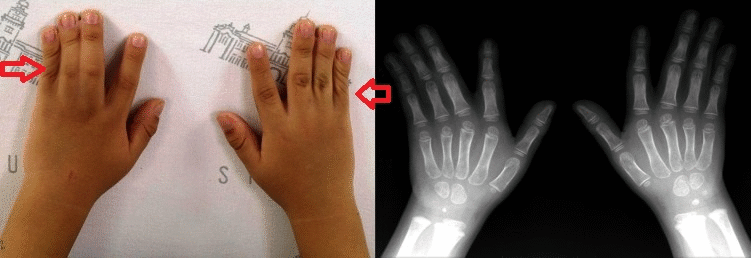
Bilateral cutaneous syndactyly of the third, fourth, and fifth fingers. Case courtesy by Hyo Soon An Et Al[41]
.jpg)

_.gif)
Physical Examination

HEENT
- HEENT examination of patients with Timothy syndrome is usually shows characteristic Craniofacial features such as:[43][44]
- Bald head
- Depressed nasal bridge
- Premaxillary underdevelopment
- Low-set ears
- Thin vermilion border of the upper lip
- Round face
- Poor dental enamel with widely spaced teeth
Heart
- Cardiovascular manifestations of Timothy Syndrome may include:[45]
- Functional 2:1 AV block
- Tachyarrhythmias
- Signs of congenital heart disease which include: PDA(Patent ductus arteriosus), PFO(patent foramen ovale), VSD(Ventricular septal defect), TOF(Tetralogy of Fallot), HCM(Hypertrophic cardiomyopathy)
Neurodevelopmental
- Neurodevelopmental examination of patients with Timothy syndrome shows signs of the following:[46][47]
- Autism
- Autism spectrum disorder
- Severe language delay
- Mild language delay
Extremities
- Extremities examination of patients with Timothy syndrome shows webbed fingers(syndactyly) and toes.[48][49][50][28][29]
- Syndactyly is due to the failure of apoptosis on the apical ectodermal ridge.
Laboratory Findings
- There are no diagnostic laboratory findings associated with Timothy syndrome.
Ultrasound
- Ultrasound may be helpful in the diagnosis of syndactyly during pregnancy with Timothy syndrome patients.
X Ray, CT scan and MRI scan
Medical Therapy
- Verapamil, mexiletine and ranolazine are also effective in treating the patients of Timothy syndrome.[51][52][53][54][55]
- Beta-blockers helps in controlling the QT interval prolongation which in-turn helps in preventing ventricular tachycardia, which is the main cause of death in patients with Timothy syndrome.
- In patients with Timothy syndrome despite treated with the beta-blockers risk of cardiac events still persists.[56]
- Preferred regimen (1): Nadolol 1–1.5 mg/kg/day administered once a day in patients ≥12 years of age, divided twice daily in infants and children
- Preferred regimen (2): Verapamil 120 mg twice a day and by decreasing the beta blocker dosage to half.
Interventions
- The feasibility of interventions depends on the severity of Timothy syndrome patients at the time of diagnosis which include:[57][58]
Implantable cardioverter-defibrillators (ICDs)
- ICDs are single most effective method for preventing sudden death in patients with Timothy syndrome.[59][60]
- ICDs are reserved for the patients who undergone cardiac arrest resuscitation.
- ICDs are good alternative choice of treatment for the patients who are resistant to beta blockers.
Left cardiac sympathetic denervation (LCSD)
- LCSDs are reserved for the patients who are not compatible with beta blocker or Implantable cardioverter-defibrillators (ICDs).
Pacemaker
- In patients with Timothy syndrome placing a pacemaker is going to help in controlling the 2:1 AV block and bradycardia.
Surgery
- Surgery is not the first-line treatment option for patients with Timothy syndrome. Surgery is usually reserved for patients with either:
- Syndactyly: Surgical release of syndactyly.[61]
Primary Prevention
- There are no established measures for the primary prevention of Timothy syndrome.
Secondary Prevention
- Effective measures for the secondary prevention of Timothy syndrome include:[62]
- Taking special care while giving the anesthesia due to the risk of cardiac arrhythmias.
- Monitoring of serum glucose levels due to intractable hypoglycemia in patients with Timothy syndrome.
References
- ↑ 1.0 1.1 Reichenbach H, Meister EM, Theile H (1992). "[The heart-hand syndrome. A new variant of disorders of heart conduction and syndactylia including osseous changes in hands and feet]". Kinderarztl Prax. 60 (2): 54–6. PMID 1318983.
- ↑ Marks ML, Trippel DL, Keating MT (1995). "Long QT syndrome associated with syndactyly identified in females". Am J Cardiol. 76 (10): 744–5. doi:10.1016/s0002-9149(99)80216-1. PMID 7572644.
- ↑ Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L; et al. (2012). "Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome". Am J Med Genet A. 158A (1): 182–7. doi:10.1002/ajmg.a.34355. PMC 3319791. PMID 22106044.
- ↑ Schultz D, Mikala G, Yatani A, Engle DB, Iles DE, Segers B; et al. (1993). "Cloning, chromosomal localization, and functional expression of the alpha 1 subunit of the L-type voltage-dependent calcium channel from normal human heart". Proc Natl Acad Sci U S A. 90 (13): 6228–32. doi:10.1073/pnas.90.13.6228. PMC 46901. PMID 8392192.
- ↑ Hiippala A, Tallila J, Myllykangas S, Koskenvuo JW, Alastalo TP (2015). "Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development". Am J Med Genet A. 167A (3): 629–34. doi:10.1002/ajmg.a.36924. PMID 25691416.
- ↑ Soldatov NM, Bouron A, Reuter H (1995). "Different voltage-dependent inhibition by dihydropyridines of human Ca2+ channel splice variants". J Biol Chem. 270 (18): 10540–3. doi:10.1074/jbc.270.18.10540. PMID 7737988.
- ↑ Lyford GL, Strege PR, Shepard A, Ou Y, Ermilov L, Miller SM; et al. (2002). "alpha(1C) (Ca(V)1.2) L-type calcium channel mediates mechanosensitive calcium regulation". Am J Physiol Cell Physiol. 283 (3): C1001–8. doi:10.1152/ajpcell.00140.2002. PMID 12176756.
- ↑ Walsh MA, Turner C, Timothy KW, Seller N, Hares DL, James AF; et al. (2018). "A multicentre study of patients with Timothy syndrome". Europace. 20 (2): 377–385. doi:10.1093/europace/euw433. PMID 28371864.
- ↑ Baurand A, Falcon-Eicher S, Laurent G, Villain E, Bonnet C, Thauvin-Robinet C; et al. (2017). "Incomplete Timothy syndrome secondary to a mosaic mutation of the CACNA1C gene diagnosed using next-generation sequencing". Am J Med Genet A. 173 (2): 531–536. doi:10.1002/ajmg.a.38045. PMID 27868338.
- ↑ Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintrón M, Scott JD; et al. (2011). "Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein". Circ Res. 109 (3): 255–61. doi:10.1161/CIRCRESAHA.111.248252. PMC 3151468. PMID 21700933.
- ↑ Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R; et al. (2004). "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell. 119 (1): 19–31. doi:10.1016/j.cell.2004.09.011. PMID 15454078.
- ↑ Dixon RE, Cheng EP, Mercado JL, Santana LF (2012). "L-type Ca2+ channel function during Timothy syndrome". Trends Cardiovasc Med. 22 (3): 72–6. doi:10.1016/j.tcm.2012.06.015. PMC 3640256. PMID 22999068.
- ↑ Dick IE, Joshi-Mukherjee R, Yang W, Yue DT (2016). "Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca(2+)-dependent inactivation". Nat Commun. 7: 10370. doi:10.1038/ncomms10370. PMC 4740114. PMID 26822303.
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ De Oliveira CC, Figueiredo EA, Gazzinelli G, Howells RE, Pellegrino J (1975). "Biochemical changes in the transformation of Schistosoma mansoni cercariae to schistosomules". Comp Biochem Physiol B. 51 (4): 417–20. doi:10.1016/0305-0491(75)90031-0. PMID 1149428.
- ↑ Betzenhauser MJ, Pitt GS, Antzelevitch C (2015). "Calcium Channel Mutations in Cardiac Arrhythmia Syndromes". Curr Mol Pharmacol. 8 (2): 133–42. doi:10.2174/1874467208666150518114857. PMC 4762596. PMID 25981977.
- ↑ Corona-Rivera JR, Barrios-Prieto E, Nieto-García R, Bloise R, Priori S, Napolitano C; et al. (2015). "Unusual retrospective prenatal findings in a male newborn with Timothy syndrome type 1". Eur J Med Genet. 58 (6–7): 332–5. doi:10.1016/j.ejmg.2015.04.001. PMID 25882468.
- ↑ Boczek NJ, Miller EM, Ye D, Nesterenko VV, Tester DJ, Antzelevitch C; et al. (2015). "Novel Timothy syndrome mutation leading to increase in CACNA1C window current". Heart Rhythm. 12 (1): 211–9. doi:10.1016/j.hrthm.2014.09.051. PMC 4907369. PMID 25260352.
- ↑ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K; et al. (1993). "GeneReviews®". PMID 20301308.
- ↑ Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC; et al. (2001). "Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome". JAMA. 286 (18): 2264–9. doi:10.1001/jama.286.18.2264. PMID 11710892.
- ↑ Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C; et al. (2007). "Prevalence of long-QT syndrome gene variants in sudden infant death syndrome". Circulation. 115 (3): 361–7. doi:10.1161/CIRCULATIONAHA.106.658021. PMID 17210839.
- ↑ Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C; et al. (2001). "Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias". Circulation. 103 (1): 89–95. doi:10.1161/01.cir.103.1.89. PMID 11136691.
- ↑ Wedekind H, Bajanowski T, Friederich P, Breithardt G, Wülfing T, Siebrands C; et al. (2006). "Sudden infant death syndrome and long QT syndrome: an epidemiological and genetic study". Int J Legal Med. 120 (3): 129–37. doi:10.1007/s00414-005-0019-0. PMID 16012827.
- ↑ Juang JJ, Horie M (2016). "Genetics of Brugada syndrome". J Arrhythm. 32 (5): 418–425. doi:10.1016/j.joa.2016.07.012. PMC 5063259. PMID 27761167.
- ↑ Thomas D, Wimmer AB, Karle CA, Licka M, Alter M, Khalil M; et al. (2005). "Dominant-negative I(Ks) suppression by KCNQ1-deltaF339 potassium channels linked to Romano-Ward syndrome". Cardiovasc Res. 67 (3): 487–97. doi:10.1016/j.cardiores.2005.05.003. PMID 15950200.
- ↑ Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L; et al. (2012). "Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome". Am J Med Genet A. 158A (1): 182–7. doi:10.1002/ajmg.a.34355. PMC 3319791. PMID 22106044.
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ 28.0 28.1 Splawski I, Timothy K, Sharpe L, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz P, Joseph R, Condouris K, Tager-Flusberg H, Priori S, Sanguinetti M, Keating M (2004). "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell. 119 (1): 19–31. PMID 15454078.
- ↑ 29.0 29.1 Splawski I, Timothy K, Decher N, Kumar P, Sachse F, Beggs A, Sanguinetti M, Keating M (2005). "Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations". Proc Natl Acad Sci U S A. 102 (23): 8089–96, discussion 8086-8. PMID 15863612.
- ↑ Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L; et al. (2012). "Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome". Am J Med Genet A. 158A (1): 182–7. doi:10.1002/ajmg.a.34355. PMC 3319791. PMID 22106044.
- ↑ Tester DJ, Ackerman MJ (2014). "Genetics of long QT syndrome". Methodist Debakey Cardiovasc J. 10 (1): 29–33. doi:10.14797/mdcj-10-1-29. PMC 4051331. PMID 24932360.
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ Tester DJ, Ackerman MJ (2014). "Genetics of long QT syndrome". Methodist Debakey Cardiovasc J. 10 (1): 29–33. doi:10.14797/mdcj-10-1-29. PMC 4051331. PMID 24932360.
- ↑ Hiippala A, Tallila J, Myllykangas S, Koskenvuo JW, Alastalo TP (2015). "Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development". Am J Med Genet A. 167A (3): 629–34. doi:10.1002/ajmg.a.36924. PMID 25691416.
- ↑ "A rare association of long QT syndrome and syndactyly: Timothy Syndrome (LQT 8)".
- ↑ Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S; et al. (2011). "Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals". J Am Coll Cardiol. 57 (1): 51–9. doi:10.1016/j.jacc.2010.07.038. PMC 3332533. PMID 21185501.
- ↑ Marks ML, Whisler SL, Clericuzio C, Keating M (1995). "A new form of long QT syndrome associated with syndactyly". J Am Coll Cardiol. 25 (1): 59–64. doi:10.1016/0735-1097(94)00318-k. PMID 7798527.
- ↑ Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH; et al. (2005). "Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations". Proc Natl Acad Sci U S A. 102 (23): 8089–96, discussion 8086-8. doi:10.1073/pnas.0502506102. PMC 1149428. PMID 15863612.
- ↑ "TRPM4 non-selective cation channel variants in long QT syndrome" Check
|url=value (help). - ↑ Tester DJ, Ackerman MJ (2014). "Genetics of long QT syndrome". Methodist Debakey Cardiovasc J. 10 (1): 29–33. doi:10.14797/mdcj-10-1-29. PMC 4051331. PMID 24932360.
- ↑ "Sudden Cardiac Arrest during Anesthesia in a 30-Month-Old Boy with Syndactyly: A Case of Genetically Proven Timothy Syndrome".
- ↑ "A rare association of long QT syndrome and syndactyly: Timothy Syndrome (LQT 8)".
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K; et al. (1993). "GeneReviews®". PMID 20301577.
- ↑ Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R; et al. (2004). "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell. 119 (1): 19–31. doi:10.1016/j.cell.2004.09.011. PMID 15454078.
- ↑ Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R; et al. (2004). "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell. 119 (1): 19–31. doi:10.1016/j.cell.2004.09.011. PMID 15454078.
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ An HS, Choi EY, Kwon BS, Kim GB, Bae EJ, Noh CI; et al. (2013). "Sudden cardiac arrest during anesthesia in a 30-month-old boy with syndactyly: a case of genetically proven Timothy syndrome". J Korean Med Sci. 28 (5): 788–91. doi:10.3346/jkms.2013.28.5.788. PMC 3653096. PMID 23678275.
- ↑ Marks M, Whisler S, Clericuzio C, Keating M (1995). "A new form of long QT syndrome associated with syndactyly". J Am Coll Cardiol. 25 (1): 59–64. PMID 7798527.
- ↑ Marks M, Trippel D, Keating M (1995). "Long QT syndrome associated with syndactyly identified in females". Am J Cardiol. 76 (10): 744–5. PMID 7572644.
- ↑ Jacobs, Avrum; Knight, Bradley P.; McDonald, Karen T.; Burke, Martin C. (2006). "Verapamil decreases ventricular tachyarrhythmias in a patient with Timothy syndrome (LQT8)". Heart Rhythm. 3 (8): 967–970. doi:10.1016/j.hrthm.2006.04.024. ISSN 1547-5271.
- ↑ Shah DP, Baez-Escudero JL, Weisberg IL, Beshai JF, Burke MC (2012). "Ranolazine safely decreases ventricular and atrial fibrillation in Timothy syndrome (LQT8)". Pacing Clin Electrophysiol. 35 (3): e62–4. doi:10.1111/j.1540-8159.2010.02913.x. PMID 20883512.
- ↑ Gao Y, Xue X, Hu D, Liu W, Yuan Y, Sun H; et al. (2013). "Inhibition of late sodium current by mexiletine: a novel pharmotherapeutical approach in timothy syndrome". Circ Arrhythm Electrophysiol. 6 (3): 614–22. doi:10.1161/CIRCEP.113.000092. PMID 23580742.
- ↑ Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J; et al. (2017). "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". Am J Med Genet A. 173 (3): 784–789. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ Gao Y, Xue X, Hu D, Liu W, Yuan Y, Sun H; et al. (2013). "Inhibition of late sodium current by mexiletine: a novel pharmotherapeutical approach in timothy syndrome". Circ Arrhythm Electrophysiol. 6 (3): 614–22. doi:10.1161/CIRCEP.113.000092. PMID 23580742.
- ↑ Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C; et al. (2009). "High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment "failures"". Circulation. 119 (2): 215–21. doi:10.1161/CIRCULATIONAHA.108.772533. PMID 19118258.
- ↑ Alexander ME, Cecchin F, Walsh EP, Triedman JK, Bevilacqua LM, Berul CI (2004). "Implications of implantable cardioverter defibrillator therapy in congenital heart disease and pediatrics". J Cardiovasc Electrophysiol. 15 (1): 72–6. doi:10.1046/j.1540-8167.2004.03388.x. PMID 15028076.
- ↑ Jons C, Moss AJ, Goldenberg I, Liu J, McNitt S, Zareba W; et al. (2010). "Risk of fatal arrhythmic events in long QT syndrome patients after syncope". J Am Coll Cardiol. 55 (8): 783–8. doi:10.1016/j.jacc.2009.11.042. PMID 20170817.
- ↑ Corona-Rivera JR, Barrios-Prieto E, Nieto-García R, Bloise R, Priori S, Napolitano C; et al. (2015). "Unusual retrospective prenatal findings in a male newborn with Timothy syndrome type 1". Eur J Med Genet. 58 (6–7): 332–5. doi:10.1016/j.ejmg.2015.04.001. PMID 25882468.
- ↑ Hiippala A, Tallila J, Myllykangas S, Koskenvuo JW, Alastalo TP (2015). "Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development". Am J Med Genet A. 167A (3): 629–34. doi:10.1002/ajmg.a.36924. PMID 25691416.
- ↑ Braun TL, Trost JG, Pederson WC (2016). "Syndactyly Release". Semin Plast Surg. 30 (4): 162–170. doi:10.1055/s-0036-1593478. PMC 5115922. PMID 27895538.
- ↑ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K; et al. (1993). "GeneReviews®". PMID 20301579.