Sandbox simrat
|
Delusional disorder Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Sandbox simrat On the Web |
|
American Roentgen Ray Society Images of Sandbox simrat |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]Associate Editor(s)-in-Chief: Simrat Sarai, M.D. [2]
Overview
The incidence of delusional disorders is approximately 0.7 to 3.0 cases per 100, 000 individuals annually. The prevalence of delusional disorders is approximately 24 to 30 cases per 100, 000 individuals annually.
Epidemiology and Demographics
Delusional disorder accounts for approximately 1-2% of admissions to inpatient mental health facilities. The incidence of first admissions for delusional disorder is approximately 0.001-0.003%. The lifetime morbid risk of delusional disorder has been estimated to range from 0.05 to 0.1 percent. According to DSM-5, the estimated lifetime prevalence of delusional disorder is 0.2 percent, which is far lower than the estimated lifetime prevalence for other major psychotic disorders such as schizophrenia and bipolar I disorder with lifetime prevalence of 0.3 to 0.87 percent and 0.24 to 0.6 percent respectively. Approximate rates of delusional disorder reported in samples of patients receiving mental health treatment have ranged from 0.5 to 1.2 percent. As the majority of people with delusional disorder do not regard it as an illness or receive treatment, studies likely underestimate the prevalence of the disorder. Delusional disorders are uncommon in psychiatric practice, though this may be an underestimation due to the fact that those afflicted lack insight and thus avoid psychiatric assessment.
Incidence
The incidence of delusional disorders is approximately 0.7 to 3.0 cases per 100, 000 individuals annually.
Prevalence
The prevalence of delusional disorders is approximately 24 to 30 cases per 100, 000 individuals annually.
Gender
Females are more commonly affected with delusional disorder than males.
Age
Delusional disorder commonly affects individuals in middle to late adult life. First admissions to hospital for delusional disorder occur between age 33 and 55 years of age.
References
- Other possible risk factors
The following factors have been linked with salivary gland cancer, but there is not enough evidence to show they are known risk factors. Further study is needed to clarify the role of these factors for salivary gland cancer.
- Epstein-Barr virus (EBV)
- Some studies have shown a link between EBV infection and a type of salivary gland cancer called lymphoepithelial carcinoma. This type of cancer is mostly seen in Inuit and people living in southern China.
- Human papillomavirus
- Workplace exposure to certain substances
- People who are involved in rubber manufacturing, asbestos mining and plumbing have an increased risk of salivary gland cancer.
- Tobacco and alcohol
- While tobacco is a strong risk factor for other types of head and neck cancer, the evidence isn’t as strong for salivary gland cancer. Some studies show that people who use tobacco products have a higher risk, while others don’t show the same results. The risk of developing salivary gland cancer from using smokeless tobacco, such as chewing tobacco or snuff, is not very clear. Although tobacco and alcohol can increase the risk for several cancers of the head and neck area, they have not been strongly linked to salivary gland cancers in most studies.
- Diet
- Some studies have found that a diet high in animal fat and low in vegetables may increase the risk of salivary gland cancer, but more research is needed to confirm this possible link.
- Cell phone use
- One study has suggested an increased risk of parotid gland tumors among heavy cell phone users. In this study, most of the tumors seen were benign. Other studies looking at this issue have not found such a link. Research in this area is still in progress.
Stage I Salivary Gland Cancer
Treatment for stage I salivary gland cancer depends on whether the cancer is low-grade or high-grade. If the cancer is low-grade, treatment may include the following:
- Surgery with or without radiation therapy.
- Fast neutron radiation therapy.
If the cancer is high-grade, treatment may include the following:
- Surgery with or without radiation therapy.
- A clinical trial of chemotherapy.
- A clinical trial of a new local therapy.
Stage II Salivary Gland Cancer
Treatment for stage II salivary gland cancer depends on whether the cancer is low-grade (slow growing) or high-grade (fast growing). If the cancer is low-grade, treatment may include the following:
- Surgery with or without radiation therapy.
- Radiation therapy.
- Chemotherapy.
If the cancer is high-grade, treatment may include the following:
- Surgery with or without radiation therapy.
- Fast neutron or photon-beam radiation therapy.
- A clinical trial of radiation therapy and/or radiosensitizers.
- A clinical trial of chemotherapy.
Stage III Salivary Gland Cancer
Treatment for stage III salivary gland cancer depends on whether the cancer is low-grade (slow growing) or high-grade (fast growing). If the cancer is low-grade, treatment may include the following:
- Surgery with or without lymphadenectomy. Radiation therapy may also be given after surgery.
- Radiation therapy
- Fast neutron radiation therapy to lymph nodes with cancer
- Chemotherapy
If the cancer is high-grade, treatment may include the following:
- Surgery with or without lymphadenectomy. Radiation therapy may also be given after surgery.
- Fast neutron radiation therapy.
- Radiation therapy as palliative therapy to relieve symptoms and improve quality of life.
- A clinical trial of radiation therapy and/or radiosensitizers.
- A clinical trial of chemotherapy.
Stage IV Salivary Gland Cancer
Treatment of stage IV salivary gland cancer may include the following:
- Fast neutron or photon-beam radiation therapy.
- A clinical trial of chemotherapy with or without radiation therapy.
Histopathological findings on biopsy can be found here.
- Regular health checkups are recommended if symptoms of salivary gland cancer develop. As salivary gland cancer is not common, so doctors do not recommend testing for it unless someone has developed symptoms. Still, in many cases salivary gland cancer can be found early, because of its location. Often patients, or their doctors may notice a lump within one of the salivary glands. The routine part of general medical and dental check-ups include checking the salivary glands for tumors.
- Physical examination should document size of the mass, mobility of the mass, fixation of the mass to the overlying skin or to the deep structures, pain with palpation, any limitation in jaw opening, buccal involvement or pharyngeal asymmetry, skin or scalp lesions indicative of primary malignancy.
- Tumors of a major salivary gland typically present with a painless mass or swelling of the submandibular, parotid, or sublingual gland.
- Tumors of the minor salivary gland arising within the oral cavity may present with a painless submucosal mass or mucosal ulceration in the palate, lips, or buccal mucosa, with an appearance similar to sialometaplasia or squamous cell carcinoma.
- The clinical presentation of a salivary gland neoplasm depends upon its specific site of origin and the extent of involvement of adjacent organs. The most common symptom of major salivary gland cancer is a painless lump in the affected gland, sometimes accompanied by paralysis of the facial nerve. Symptoms due to more advanced minor salivary gland tumors are a function of the location of the tumor and can include nasal obstruction, congestion, vision changes, or trismus when present in the nasal cavity or maxillary sinus. Minor salivary gland tumors involving the nasopharynx usually present at an advanced stage; invasion of the skull base, intracranial extension, or involvement of cranial nerves is common.[4]
- Persistent facial pain is highly suggestive of malignancy. Approximately 10% to 15% of malignant parotid neoplasma present with pain.
- Neurological signs, such as numbness or weakness caused by nerve involvement, typically indicate a malignancy.
. The white area in the upper lobe is cancer; the black areas indicate the patient was a smoker]]
Genetic deficiencies and a rare autoimmune disorder may lower plasma clotting factor levels needed for a normal clotting process. When a blood vessel is injured, a temporary scab does form, but the missing coagulation factors prevent fibrin formation which is necessary to maintain the blood clot. Therefore, there is no increase in bleeding time with haemophilia because platelets are intact, allowing the formation of these temporary hemostatic plugs (clots). However, "late" bleeding is affected, because these hemostatic plugs are not able to be maintained.
===Genetic structure===800px-Classical_blood_coagulation_pathway.png

Females possess two X-chromosomes, whereas males have one X and one Y chromosome. Since the mutations causing the disease are recessive, a woman carrying the defect on one of her X-chromosomes may not be affected, as the equivalent allele on her other chromosome should express itself to produce the necessary clotting factors. However the Y-chromosome in men has no gene for factors VIII or IX.
If the genes responsible for production of factor VIII or factor IX present on a male's X-chromosome is deficient then there is no equivalent on the Y-chromosome, so the deficient gene is not masked by the dominant allele and he will develop the disease.
Since a male receives his single X-chromosome from his mother, the son of a healthy female silently carrying the deficient gene will have a 50% chance of inheriting that gene from her and with it the disease; and if his mother is affected with hemophilia, he will have a 100% chance of being a haemophiliac.
In contrast, for a female to inherit the disease, she must receive two deficient X-chromosomes, one from her mother and the other from her father (who must therefore be a hemophiliac himself). Hence hemophilia is far more common among males than females. However it is possible for female carriers to become mild hemophiliacs due to lyonisation of the X chromosomes.
Hemophiliac females are more common than they once were, as improved treatments for the disease have allowed more hemophiliac males to survive to adulthood and become parents. Adult females may experience menorrhagia (heavy periods) due to the bleeding tendency. The pattern of inheritance is criss-cross type. This type of pattern is also seen in color blindness.
As with all genetic disorders, it is also possible for a human to acquire it spontaneously (de novo), rather than inheriting it, because of a new mutation in one of their parents' gametes. Spontaneous mutations account for about ⅓ of all hemophilia A and 20% of all hemophilia B cases.
If a female gives birth to a hemophiliac child, either the female is a carrier for the disease or the hemophilia was the result of a spontaneous mutation. Until modern direct DNA testing, it was impossible to determine if a female with only healthy children was a carrier or not. Generally, the more healthy sons she bore, the higher the probability that she was not a carrier. If the RH factor of the born male is different from the mother, the child will not be affected.
If a male is afflicted with the disease and has children, his daughters will be carriers for hemophilia. His sons, however, will not be affected with the disease. This is because the disease is X-linked and the father can not pass hemophilia through the Y chromosome.
Factor VIII production, processing and structure
FVIII is a glycoprotein procofactor. Although the primary site of release in humans is ambiguous, it is synthesized and released into the bloodstream by the vascular, glomerular, and tubular endothelium, and the sinusoidal cells of the liver. Hemophilia A has been corrected by liver transplantation. Transplanting hepatocytes was ineffective, but liver endothelial cells were effective.In the blood, it mainly circulates in a stable noncovalent complex with von Willebrand factor. Upon activation by thrombin, (factor IIa), it dissociates from the complex to interact with factor IXa in the coagulation cascade. It is a cofactor to factor IXa in the activation of factor X, which, in turn, with its cofactor factor Va, activates more thrombin. Thrombin cleaves fibrinogen into fibrin which polymerizes and crosslinks (using factor XIII) into a blood clot. No longer protected by vWF, activated FVIII is proteolytically inactivated in the process (most prominently by activated protein C and factor IXa) and quickly cleared from the blood stream.
Factor VIII is not affected by liver disease. In fact, levels usually are elevated in such instances.
Von Willebrand Factor[vWF] synthesis, structure and function
vWF is a large multimeric glycoprotein present in blood plasma and produced constitutively as ultra-large vWF in endothelium (in the Weibel-Palade bodies), megakaryocytes (α-granules of platelets), and subendothelial connective tissue.The basic vWF monomer is a 2050-amino acid protein. Every monomer contains a number of specific domains with a specific function. Von Willebrand factor primary function is binding to other proteins, in particular factor VIII, and it is important in platelet adhesion to wound sites. It is not an enzyme and, thus, has no catalytic activity. vWF binds to a number of cells and molecules. The most important ones are:
- Factor VIII is bound to vWF while inactive in circulation; factor VIII degrades rapidly when not bound to vWF. Factor VIII is released from vWF by the action of thrombin.
- vWF binds to collagen, e.g., when it is exposed in endothelial cells due to damage occurring to the blood vessel.
- vWF binds to platelet gpIb when it forms a complex with gpIX and gpV; this binding occurs under all circumstances, but is most efficient under high shear stress (i.e., rapid blood flow in narrow blood vessels, see below).
- vWF binds to other platelet receptors when they are activated, e.g., by thrombin (i.e., when coagulation has been stimulated).
vWF plays a major role in blood coagulation. Therefore, vWF deficiency or dysfunction (von Willebrand disease) leads to a bleeding tendency, which is most apparent in tissues having high blood flow shear in narrow vessels. From studies it appears that vWF uncoils under these circumstances, decelerating passing platelets. Calcium enhances the refolding rate of vWF A2 domain, allowing the protein to act as a shear force sensor.
Factor IX synthesis, structure and function
- Factor IX (or Christmas factor) is one of the serine proteases of the coagulation system; it belongs to peptidase family S1. Deficiency of this protein causes hemophilia B. Factors VII, IX, and X all play key roles in blood coagulation and also share a common domain architecture. The factor IX protein is composed of four protein domains: the Gla domain, two tandem copies of the EGF domain and a C-terminal trypsin-like peptidase domain which carries out the catalytic cleavage.The N-terminal EGF domain has been shown to at least in part be responsible for binding tissue factor. Wilkinson et al. conclude that residues 88 to 109 of the second EGF domain mediate binding to platelets and assembly of the factor X activating complex. The structures of all four domains have been solved. A structure of the two EGF domains and the trypsin-like domain was determined for the pig protein. The structure of the Gla domain, which is responsible for Ca(II)-dependent phospholipid binding, was also determined by NMR. Several structures of 'super active' mutants have been solved, which reveal the nature of factor IX activation by other proteins in the clotting cascade.
- Factor IX is produced as a zymogen, an inactive precursor. It is processed to remove the signal peptide, glycosylated and then cleaved by factor XIa (of the contact pathway) or factor VIIa (of the tissue factor pathway) to produce a two-chain form where the chains are linked by a disulfide bridge. When activated into factor IXa, in the presence of Ca2+, membrane phospholipids, and a Factor VIII cofactor, it hydrolyses one arginine-isoleucine bond in factor X to form factor Xa. Factor IX is inhibited by antithrombin. Factor IX expression increases with age in humans and mice. In mouse models mutations within the promoter region of factor IX have an age-dependent phenotype.
Clotting cascade

The coagulation cascade of secondary hemostasis has two initial pathways which lead to fibrin formation. These are the contact activation pathway (also known as the intrinsic pathway), and the tissue factor pathway (also known as the extrinsic pathway) which both lead to the same fundamental reactions that produce fibrin. It was previously thought that the two pathways of coagulation cascade were of equal importance, but it is now known that the primary pathway for the initiation of blood coagulation is the tissue factor pathway. The pathways are a series of reactions, in which a zymogen (inactive enzyme precursor) of a serine protease and its glycoprotein co-factor are activated to become active components that then catalyze the next reaction in the cascade, ultimately resulting in cross-linked fibrin. Coagulation factors are generally indicated by Roman numerals, with a lowercase a appended to indicate an active form. The coagulation factors are generally serine proteases (enzymes), which act by cleaving downstream proteins. There are some exceptions. For example, FVIII and FV are glycoproteins, and Factor XIII is a transglutaminase. The coagulation factors circulate as inactive zymogens. The coagulation cascade is therefore classically divided into three pathways. The tissue factor and contact activation pathways both activate the "final common pathway" of factor X, thrombin and fibrin.
Tissue factor pathway (extrinsic)
The main role of the tissue factor pathway is to generate a "thrombin burst", a process by which thrombin, the most important constituent of the coagulation cascade in terms of its feedback activation roles, is released very rapidly. FVIIa circulates in a higher amount than any other activated coagulation factor. The process includes the following steps:
- Following damage to the blood vessel, FVII leaves the circulation and comes into contact with tissue factor (TF) expressed on tissue-factor-bearing cells (stromal fibroblasts and leukocytes), forming an activated complex (TF-FVIIa).
- TF-FVIIa activates FIX and FX.
- FVII is itself activated by thrombin, FXIa, FXII and FXa.
- The activation of FX (to form FXa) by TF-FVIIa is almost immediately inhibited by tissue factor pathway inhibitor (TFPI).
- FXa and its co-factor FVa form the prothrombinase complex, which activates prothrombin to thrombin.
- Thrombin then activates other components of the coagulation cascade, including FV and FVIII (which activates FXI, which, in turn, activates FIX), and activates and releases FVIII from being bound to vWF.
- FVIIa is the co-factor of FIXa, and together they form the "tenase" complex, which activates FX; and so the cycle continues. ("Tenase" is a contraction of "ten" and the suffix "-ase" used for enzymes.)
Contact activation pathway (intrinsic)
The contact activation pathway begins with formation of the primary complex on collagen by high-molecular-weight kininogen (HMWK),prekallikrein, and FXII (Hageman factor). Prekallikrein is converted to kallikrein and FXII becomes FXIIa. FXIIa converts FXI into FXIa. Factor XIa activates FIX, which with its co-factor FVIIIa form the tenase complex, which activates FX to FXa. The minor role that the contact activation pathway has in initiating clot formation can be illustrated by the fact that patients with severe deficiencies of FXII, HMWK, and prekallikrein do not have a bleeding disorder. Instead, contact activation system seems to be more involved in inflammation.[7]
Final common pathway
The division of coagulation in two pathways is mainly artificial, it originates from laboratory tests in which clotting times were measured after the clotting was initiated by glass (intrinsic pathway) or by thromboplastin (a mix of tissue factor and phospholipids). In fact thrombin is present from the very beginning, already when platelets are making the plug.Thrombin has a large array of functions, not only the conversion of fibrinogen to fibrin, the building block of a hemostatic plug. In addition, it is the most important platelet activator and on top of that it activates Factors VIII and V and their inhibitor protein C (in the presence of thrombomodulin), and it activates Factor XIII, which forms covalent bonds that crosslink the fibrin polymers that form from activated monomers.
Following activation by the contact factor or tissue factor pathways, the coagulation cascade is maintained in a prothrombotic state by the continued activation of FVIII and FIX to form the tenase complex, until it is down-regulated by the anticoagulant pathways Dr. Sheraz was born in twin city of Islamabad, Pakistan. He graduated from Army Medical College in 2013 and did his internship in Social Security Hospital Islamabad. He has written various research articles which were published in Journal of Pakistan Medical association, International Journal of archives, Journal of physicians and surgeons Pakistan and Journal of American College of Gastroenterology.
| Condition | Prothrombin time | Partial Thromboplastin Time | Bleeding Time | Platelet Count |
|---|---|---|---|---|
| Heamophilia A or B | Unaffected | Prolonged | Unaffected | Unaffected |
| Von Willebrand Disease | Unaffected | Prolonged or Unaffected | Prolonged | Unaffected |
| Thrombocytopenia | Unaffected | Unaffected | Prolonged | Decreased |
| Adapted from Wikipedia hemophilia Laboratory Finding> "Wikipedia Hemophilia Laboratory Finding". | ||||
In the United States, most people with hemophilia are diagnosed at a very young age. The median age at diagnosis is 36 months for people with mild hemophilia, 8 months for those with moderate hemophilia, and 1 month for those with severe hemophilia. In about two thirds of cases, there is a family history of hemophilia. The diagnosis of hemophilia is made using a special blood test and most babies can be tested soon after birth. Sometimes prenatal genetic testing is done to diagnose hemophilia before birth. For the one-third of babies born with hemophilia in families with no known history of hemophilia, the diagnosis is made when an unusual bleeding event occurs. Special blood tests are required to make the diagnosis.
References
- ↑ Hafed, Layla; Farag, Heba; Shaker, Olfat; El-Rouby, Dalia (2012). "Is human papilloma virus associated with salivary gland neoplasms? An in situ-hybridization study". Archives of Oral Biology. 57 (9): 1194–1199. doi:10.1016/j.archoralbio.2012.03.009. ISSN 0003-9969.
- ↑ Boland, Jennifer M; McPhail, Ellen D; García, Joaquín J; Lewis, Jean E; Schembri-Wismayer, David J (2011). "Detection of human papilloma virus and p16 expression in high-grade adenoid cystic carcinoma of the head and neck". Modern Pathology. 25 (4): 529–536. doi:10.1038/modpathol.2011.186. ISSN 0893-3952.
- ↑ Jour, G.; West, K.; Ghali, V.; Shank, D.; Ephrem, G.; Wenig, B. M. (2013). "Differential Expression of p16INK4A and Cyclin D1 in Benign and Malignant Salivary Gland Tumors: A Study of 44 Cases". Head and Neck Pathology. 7 (3): 224–231. doi:10.1007/s12105-012-0417-9. ISSN 1936-055X.
- ↑ Salivary gland cancer. National cancer institute(2015)http://www.cancer.gov/types/head-and-neck/hp/salivary-gland-treatment-pdq#link/_410_toc Accessed on November 8, 2015
The true incidence of Acoustic neuromas has been difficult to estimate.[1] [2] The incidence has been reported to range from 1 to 20 per million per year.[3] [4] [5] A study of acoustic neuromas in a review of 24,000 brain MRIs reported a prevalence of 0.07%. When comparing the clinical incidence of acoustic neuromas to the prevalence of occult acoustic neuromas ascertained from histopathological studies of temporal bones, it can be concluded that the vast majority of tumors that exist are never clinically manifested.[6] [7] Acoustic neuromas are most commonly diagnosed between the ages of 30 and 68.[8] [9] Reported cases of acoustic neuroma in childhood are rare and in such patients other evidence of NF2 should be investigated. In a study of 146 cases of acoustic neuromas, the median age of cases was 52 years.[10] In a second study of 793 cases of Acoustic neuroma, the median age of the cases was 54 years. The sex ratio (females/males) for acoustic neuromas has been reported to be >1 [11] [12] [13] [14] However, data from the Central Brain Tumor Registry of the United States (CBTRUS) do not support a female/male difference[15] Nevertheless,some of the studies that analyzed Swedish data, the sex ratio (females/males) of acoustic neuromas in the Nordic counties has been shown to be >1. This sex ratio may indicate that hormones play a role in the etiology of acoustic neuromas[16] [17] The tumor has been reported to be higher in whites than in non-whites[18] as well as being uncommon in individuals of African ancestry[19] According to some studies the incidence of acoustic neuromas has been increasing. This may be due to several factors. Steady improvements in diagnosis, such as the introduction of auditory brainstem response, CT, and MRI could explain part of the increased incidence. Increased awareness among physicians and patients of the symptoms of acoustic neuromas may have caused an increase in the reporting of the tumor. Changes in classification or coding may also explain some of the trend, whereby registries may have misclassified nonvestibular schwannomas as acoustic neuromas. Or, there may be a true increase in the incidence of these tumors. The trend may also lend support to the emerging hypotheses regarding an environmental cause of these tumors. Established and hypothesized risk factors have been reported extensively in the literature and include ionizing radiation exposure, cellular telephone use, specific occupations, a possible hormonal cause, and loud noise exposure.
| Gender
|
Race
| |||||
| Type of Tumor | Total Number of Tumors (N) | Overall Rate (CI) | Male | Female | White | Nonwhite |
| CBTRUS, 1995–1999 | ||||||
| Nerve sheath | 2,811 | 1.08 (1.04–1.12) | 1.10 (1.04–1.16) | 1.07 (1.02–1.13) | 1.13 (1.08–1.17) | 0.56 (0.48–0.63) |
| Vestibular schwannoma | 1,424 | 0.55 (0.52–0.58) | 0.56 (0.52–0.60) | 0.55 (0.51–0.58) | 0.58 (0.55–0.61) | 0.23 (0.18–0.28) |
| LACCSP, 1995–1998 | ||||||
| Nerve sheath | 352 | 1.11 (0.99–1.22) | 1.15 (0.97–1.32) | 1.07 (0.91–1.23) | 1.21 (1.08–1.36) | 0.68 (0.50–0.87) |
| Vestibular schwannoma | 256 | 0.82 (0.71–0.92) | 0.83 (0.68–0.99) | (0.66–0.94)0.80 | 0.89 (0.77–1.01) | 0.51 (0.36–0.67) |
Contrast-enhanced CT will detect almost all acoustic neuromas that are greater than 2.0 cm in diameter and project further than 1.5 cm into the cerebellopontine angle. Those tumors that are smaller may be detected by MRI with gadolinium enhancement. Audiology and vestibular tests should be concurrently evaluated using air conduction and bone conduction threshold testing to assess for sensorineural versus conduction hearing loss.
MRI
| Acoustic neuromas are most frequently diagnosed by MRI scan in a patient with unilateral hearing loss. Important information to be determined from the MRI scan are distance the tumor extends laterally in the auditory canal, the extent to which the tumor expands in the cerebello-pontine angle, and whether or not the brain stem is contacted or distorted |
- The definitive diagnostic test for patients with acoustic tumors is gadolinium-enhanced MRI.
- Well-performed scanning can demonstrate tumors as small as 1-2 mm in diameter. On the other hand, thin-cut CT scanning can miss tumors as large as 1.5 cm even when intravenous contrast enhancement is used.
- Gadolinium contrast is critical because nonenhanced MRI can miss small tumors.
- Fast-spin echo techniques do not require gadolinium enhancement and can be performed very rapidly and relatively inexpensively. However, such highly targeted techniques risk missing other important causes of unilateral sensory hearing loss, including intra-axial tumors, demyelinating disease, and infarcts.
- MRI is contraindicated in individuals with ferromagnetic implants.
- Fine-cut CT scanning of the internal auditory canal with contrast can rule out a medium-size or large tumor but cannot be relied upon to detect a tumor smaller than 1-1.5 cm.
- If suspicion is high and MRI is contraindicated, air-contrast cisternography has high sensitivity and can detect relatively small intracanalicular tumors.
MRI Classification of Acoustic Neuroma:
|
Entirely intracanalicular Since these tumors are entirely within the bony canal, they pose no threat to the brain or cranial nervesother than VII and VIII, which are in the canal. Therefore, the crucial questions are how severely affected are the nerves already and how fast is the tumor growing. Since the nerve most susceptible to pressure is the auditory component of nerve VIII, a detailed hearing test and auditory evoked potentials should be performed. If useful hearing is present, it is advisable to remove the tumor or treat it with stereotactic radiation before it becomes larger. With microsurgery, the tumor can be completely removed with minimal risk and a reasonable chance of preserving hearing. Facial nerve and auditory monitoring are, of course, required. With stereotactic radiation therapy, no actual surgery is required but the tumor will still be present and will need to be followed indefinitely by MRI scans. The outcome for preservation of hearing is similar in both techniques. If useful hearing is not present, it is reasonable to follow the tumor for one year with a repeat MRI scan to see if it is growing before making a decision regarding surgery, radiation, or more waiting. However, if knowing you have a tumor in your head and not doing something is unacceptable, it is appropriate to proceed with surgery or stereotactic radiation. Intracranial extension without brainstem distortion These tumors have already demonstrated their propensity to grow, but are not yet life-threatening. They should be surgically removed if the general health of the patient permits; otherwise, they should be treated with stereotactic radiation. The surgical approach depends on the status of the hearing and the preference of the surgeon, but the primary objective other than total removal of the tumor should be to preserve the facial nerve. This requires facial nerve monitoring during the course of the operation. If useful hearing is still present, an effort should be made to preserve it by intraoperative auditory monitoring and selecting a surgical approach which does not damage the cochlea. Intracranial extension with brainstem distortion Large tumors which distort the brain stem are potentially life-threatening and should be surgically removed as soon as possible. The goal should be total removal, but great care must be taken to ensure that neither the brain stem nor its blood supply are injured in the process. Preservation of the facial nerve also remains important, and it may be necessary to leave a portion of tumor capsule to protect the brain stem, blood vessels or facial nerve. The possibility of preserving any hearing with large tumors is minimal |
-
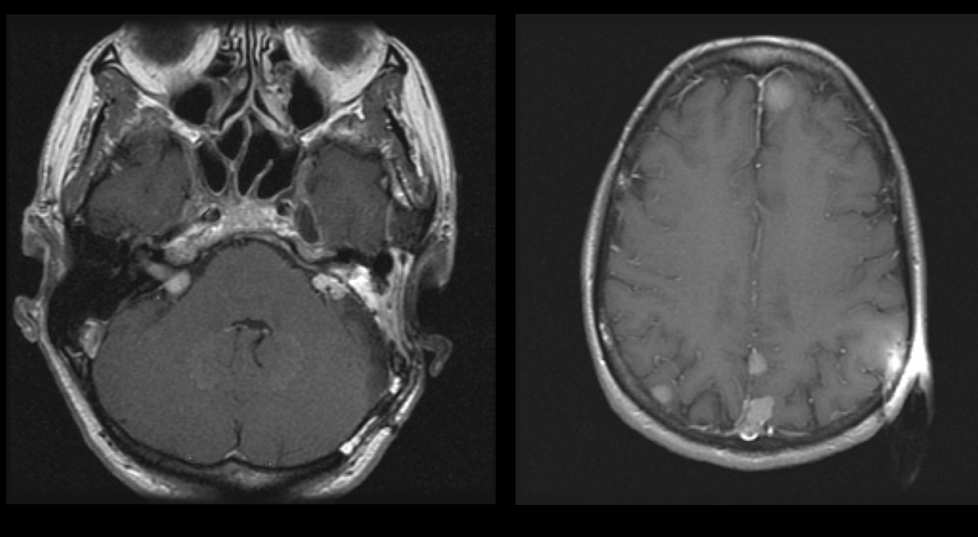 Axial image of a patient with bilateral acoustic neuromas. the patient has NF2. The right image is a higher image in the same patient showing multiple meningiomas
Axial image of a patient with bilateral acoustic neuromas. the patient has NF2. The right image is a higher image in the same patient showing multiple meningiomas -
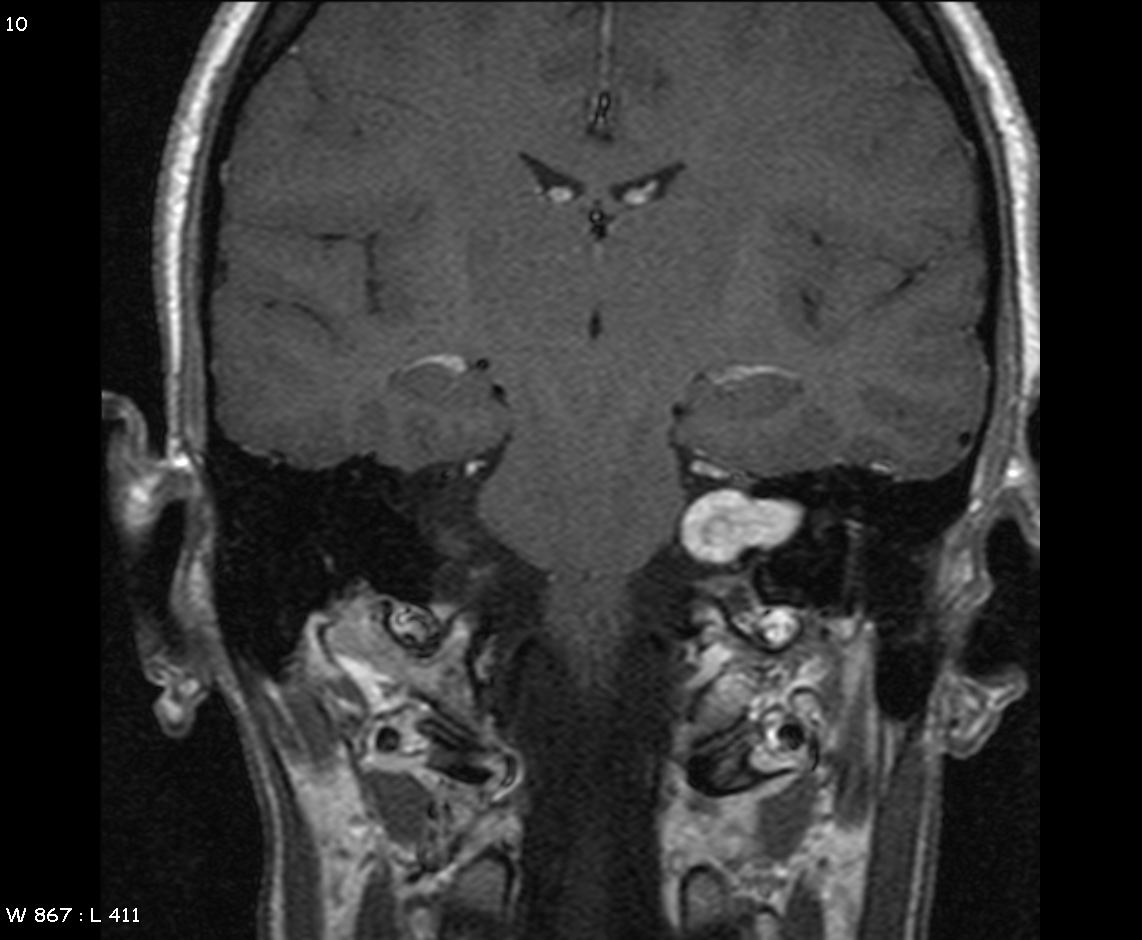 MRI demonstrating left acoustic neuroma in the CP angle==Overview==
MRI demonstrating left acoustic neuroma in the CP angle==Overview== -
Stage
-
Features
-
Stage
-
Features


.jpg){kind=link}
References
- ↑ Ho SY, Kveton JF. Acoustic neuroma assessment and management. Otolaryngol Clin North Am 2002;35:393-404
- ↑ Tos M, Charabi S, Thomsen J. Clincial experience with vestibular schwannomas: epidemiology, symptomatology, diagnosis, and surgical results. Eur Arch Otorhinolaryngol 1998;255:1-6.
- ↑ Howitz MF, Johansen C, Tos M, Charabi S, Olsen JH. Incidence of Vestibular Schwannoma in Denmark, 1977-1995. Am J Otol 2000;21:690-694
- ↑ Tos M, Charabi S, Thomsen J. Clincial experience with vestibular schwannomas: epidemiology, symptomatology, diagnosis, and surgical results. Eur Arch Otorhinolaryngol 1998;255:1-6.
- ↑ Tos M, Stangerup SE, Cayé-Thomasen P, Tos T, Thomsen J. What is the real incidence of vestibular schwannoma? Arch Otolaryngol Head Neck Surg 2004;130:216-220
- ↑ Thomsen J, Tos M. Acoustic neuroma: clinical aspects, audiovestibular assessment, diagnostic delay, and growth rate. Am J Otol 1990;11:12-19.
- ↑ Rosenberg SI. Natural history of acoustic neuromas. Laryngoscope 2000;110:497-508.
- ↑ Strasnick B, Glasscock ME, Haynes D, McMenomey SO, Minor LB. The natural history of untreated acoustic neuromas. Laryngoscope1994;104:1115-1119.
- ↑ Hart RG, Davenport J. Diagnosis of acoustic neuroma. Neurosurgery 1981;9:450-463.
- ↑ Edwards CG, Schwartzbaum JA, Lönn S, Ahlbom A, Feychting M. Exposure to loud noise and risk of acoustic neuroma. Am J Epidemiol 2006;163:327-333.
- ↑ Inskip PD, Tarone RE, Hatch EE, et al. Sociodemographic indicators and risk of brain tumours. Int J Epidemiol 2003;32:225-233.
- ↑ Howitz MF, Johansen C, Tos M, Charabi S, Olsen JH. Incidence of Vestibular Schwannoma in Denmark, 1977-1995. Am J Otol 2000;21:690-694.
- ↑ Chandler CL, Ramsden RT. Acoustic schwannoma. Br J Hosp Med 1993;49:336-343.
- ↑ Spoelhof GD. When to suspect acoustic neuroma. Am Fam Physician 1995;52:1768-1774.
- ↑ Propp JM, McCarthy BJ, Davis FG, Preston-Martin S. Descriptive epidemiology of vestibular schwannomas. Neuro-oncol 2006;8:1-11.
- ↑ Schlehofer B, Blettner M, Wahrendorf J. Association between brain tumors and menopausal status. J Natl Cancer Inst 1992;84:1346-1349
- ↑ Howitz MF, Johansen C, Tos M, Charabi S, Olsen JH. Incidence of Vestibular Schwannoma in Denmark, 1977-1995. Am J Otol 2000;21:690-694.
- ↑ Propp JM, McCarthy BJ, Davis FG, Preston-Martin S. Descriptive epidemiology of vestibular schwannomas. Neuro-oncol 2006;8:1-11.
- ↑ Chandler CL, Ramsden RT. Acoustic schwannoma. Br J Hosp Med 1993;49:336-343..