WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
LEXIVA® is indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV-1) infection.
The following points should be considered when initiating therapy with LEXIVA plus ritonavir in protease inhibitor-experienced patients:
The protease inhibitor-experienced patient trial was not large enough to reach a definitive conclusion that LEXIVA plus ritonavir and lopinavir plus ritonavir are clinically equivalent
Once-daily administration of LEXIVA plus ritonavir is not recommended for adult protease inhibitor-experienced patients or any pediatric patients
Dosing of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced pediatric patients younger than 6 months.
LEXIVA Tablets may be taken with or without food.
Adults should take LEXIVA Oral Suspension without food. Pediatric patients should take LEXIVA Oral Suspension with food . If emesis occurs within 30 minutes after dosing, re-dosing of LEXIVA Oral Suspension should occur.
Higher-than-approved dose combinations of LEXIVA plus ritonavir are not recommended due to an increased risk of transaminase elevations
When LEXIVA is used in combination with ritonavir, prescribers should consult the full prescribing information for ritonavir.
Therapy-Naive Adults:
LEXIVA 1,400 mg twice daily (without ritonavir).•LEXIVA 1,400 mg once daily plus ritonavir 200 mg once daily
LEXIVA 1,400 mg once daily plus ritonavir 100 mg once daily.∘Dosing of LEXIVA 1,400 mg once daily plus ritonavir 100 mg once daily is supported by pharmacokinetic data
LEXIVA 700 mg twice daily plus ritonavir 100 mg twice daily.∘Dosing of LEXIVA 700 mg twice daily plus 100 mg ritonavir twice daily is supported by pharmacokinetic and safety data
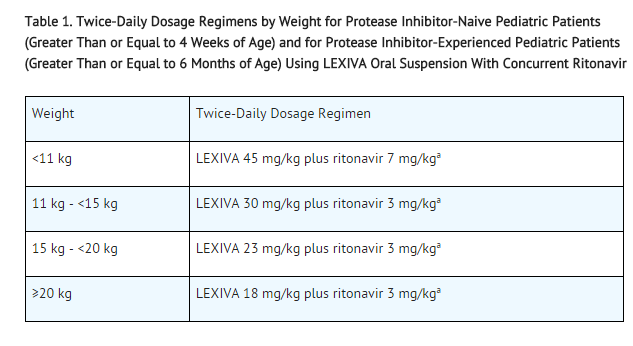
The recommended dosage of LEXIVA in patients aged at least 4 weeks to 18 years should be calculated based on body weight (kg) and should not exceed the recommended adult dose (Table 1).
Table 1. Twice-Daily Dosage Regimens by Weight for Protease Inhibitor-Naive Pediatric Patients (Greater Than or Equal to 4 Weeks of Age) and for Protease Inhibitor-Experienced Pediatric Patients (Greater Than or Equal to 6 Months of Age) Using LEXIVA Oral Suspension With Concurrent Ritonavir
This image is provided by the National Library of Medicine.
When dosing with ritonavir, do not exceed the adult dose of LEXIVA 700 mg/
ritonavir 100 mg twice-daily dose.
Alternatively, protease inhibitor-naive children aged 2 years and older can be administered LEXIVA (without ritonavir) 30 mg per kg twice daily.
LEXIVA should only be administered to infants born at 38 weeks gestation or greater and who have attained a post-natal age of 28 days.
For pediatric patients, pharmacokinetic and clinical data:
do not support once-daily dosing of LEXIVA alone or in combination with ritonavir
do not support administration of LEXIVA alone or in combination with ritonavir for protease inhibitor‑experienced children younger than 6 months
do not support twice-daily dosing of LEXIVA without ritonavir in pediatric patients younger than 2 years
Other Dosing Considerations
When administered without ritonavir, the adult regimen of LEXIVA Tablets 1,400 mg twice daily may be used for pediatric patients weighing at least 47 kg.
When administered in combination with ritonavir, LEXIVA Tablets may be used for pediatric patients weighing at least 39 kg; ritonavir capsules may be used for pediatric patients weighing at least 33 kg.
Mild Hepatic Impairment (Child-Pugh Score Ranging From 5 to 6):LEXIVA should be used with caution at a reduced dosage of 700 mg twice daily without ritonavir (therapy-naive) or 700 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
Moderate Hepatic Impairment (Child-Pugh Score Ranging From 7 to 9):LEXIVA should be used with caution at a reduced dosage of 700 mg twice daily without ritonavir (therapy-naive), or 450 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
Severe Hepatic Impairment (Child-Pugh Score Ranging From 10 to 15):LEXIVA should be used with caution at a reduced dosage of 350 mg twice daily without ritonavir (therapy-naive) or 300 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
There are no data to support dosing recommendations for pediatric patients with hepatic impairment.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Fosamprenavir in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Fosamprenavir in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Fosamprenavir in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Fosamprenavir in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Fosamprenavir in pediatric patients.
Contraindications
LEXIVA is contraindicated:
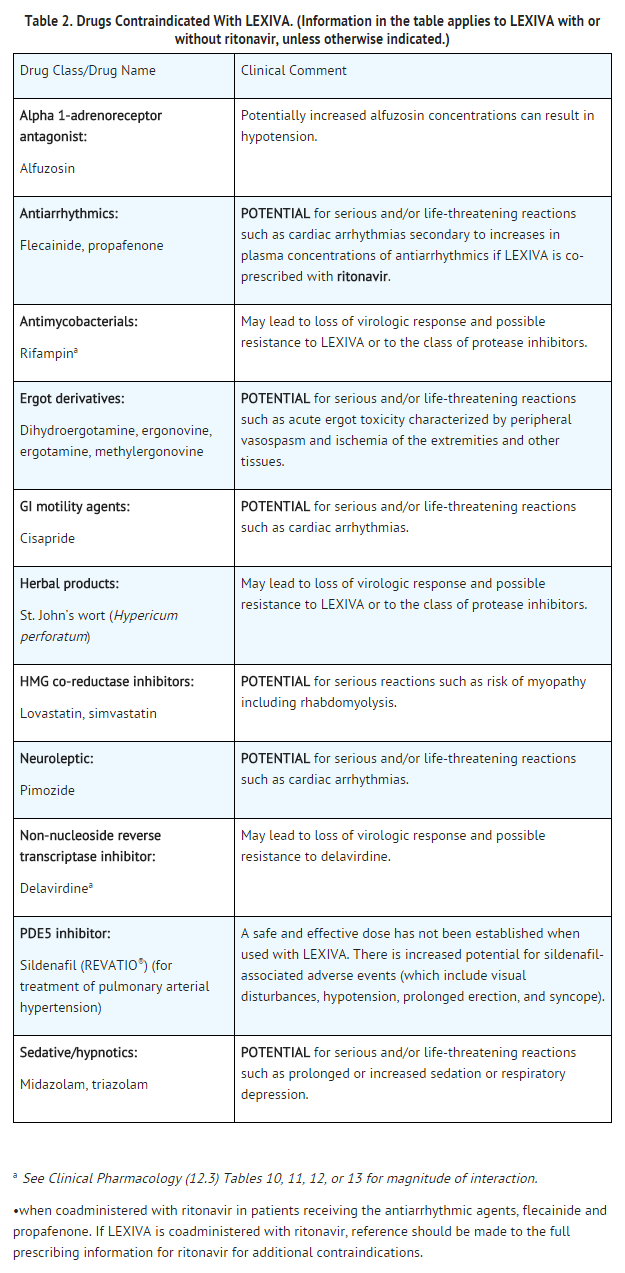
in patients with previously demonstrated clinically significant hypersensitivity (e.g., Stevens-Johnson syndrome) to any of the components of this product or to amprenavir.•when coadministered with drugs that are highly dependent on cytochrome P450 3A4 (CYP3A4) for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events (Table 2).
This image is provided by the National Library of Medicine.
when coadministered with ritonavir in patients receiving the antiarrhythmic agents, flecainide and propafenone. If LEXIVA is coadministered with ritonavir, reference should be made to the full prescribing information for ritonavir for additional contraindications.
Warnings
Certain drugs should not be coadministered with LEXIVA due to risk of serious or life-threatening adverse reactions.
LEXIVA should be discontinued for severe skin reactions including Stevens-Johnson syndrome.
LEXIVA should be used with caution in patients with a known sulfonamide allergy.
Use of higher than approved doses may lead to transaminase elevations. Patients with hepatitis B or C are at increased risk of transaminase elevations.
Patients receiving LEXIVA may develop new onset or exacerbations of diabetes mellitus, hyperglycemia, immune reconstitution syndrome, redistribution/accumulation of body fat, and elevated triglyceride and cholesterol concentrations.
Monitor cholesterol and triglycerides prior to therapy and periodically thereafter.
Acute hemolytic anemia has been reported with amprenavir.
Hemophilia: Spontaneous bleeding may occur, and additional factor VIII may be required.
Nephrolithiasis: Cases of nephrolithiasis have been reported with fosamprenavir.
See Table 2 for listings of drugs that are contraindicated due to potentially life-threatening adverse events, significant drug interactions, or loss of virologic activity.
Severe and life-threatening skin reactions, including 1 case of Stevens-Johnson syndrome among 700 subjects treated with LEXIVA in clinical trials. Treatment with LEXIVA should be discontinued for severe or life-threatening rashes and for moderate rashes accompanied by systemic symptoms.
LEXIVA should be used with caution in patients with a known sulfonamide allergy. Fosamprenavir contains a sulfonamide moiety. The potential for cross-sensitivity between drugs in the sulfonamide class and fosamprenavir is unknown. In a clinical trial of LEXIVA used as the sole protease inhibitor, rash occurred in 2 of 10 subjects (20%) with a history of sulfonamide allergy compared with 42 of 126 subjects (33%) with no history of sulfonamide allergy. In 2 clinical trials of LEXIVA plus low-dose ritonavir, rash occurred in 8 of 50 subjects (16%) with a history of sulfonamide allergy compared with 50 of 412 subjects (12%) with no history of sulfonamide allergy.
Use of LEXIVA with ritonavir at higher-than-recommended dosages may result in transaminase elevations and should not be used. Patients with underlying hepatitis B or C or marked elevations in transaminases prior to treatment may be at increased risk for developing or worsening of transaminase elevations. Appropriate laboratory testing should be conducted prior to initiating therapy with LEXIVA and patients should be monitored closely during treatment.
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-1-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and causal relationships between protease inhibitor therapy and these events have not been established.
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including LEXIVA. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance,” have been observed in patients receiving antiretroviral therapy, including LEXIVA. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Treatment with LEXIVA plus ritonavir has resulted in increases in the concentration of triglycerides and cholesterol. Triglyceride and cholesterol testing should be performed prior to initiating therapy with LEXIVA and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate.
Acute hemolytic anemia has been reported in a patient treated with amprenavir.
There have been reports of spontaneous bleeding in patients with hemophilia A and B treated with protease inhibitors. In some patients, additional factor VIII was required. In many of the reported cases, treatment with protease inhibitors was continued or restarted. A causal relationship between protease inhibitor therapy and these episodes has not been established.
Cases of nephrolithiasis were reported during postmarketing surveillance in HIV-1-infected patients receiving LEXIVA.Because these events were reported voluntarily during clinical practice, estimates of frequency cannot be made. * If signs or symptoms of nephrolithiasis occur, temporary interruption or discontinuation of therapy may be considered.
Because the potential for HIV cross-resistance among protease inhibitors has not been fully explored, it is unknown what effect therapy with LEXIVA will have on the activity of subsequently administered protease inhibitors.
LEXIVA has been studied in patients who have experienced treatment failure with protease inhibitors.
Adverse Reactions
Clinical Trials Experience
The most common moderate to severe adverse reactions in clinical trials of LEXIVA were diarrhea, rash, nausea, vomiting, and headache.
Treatment discontinuation due to adverse events occurred in 6.4% of subjects receiving LEXIVA and in 5.9% of subjects receiving comparator treatments.
The most common adverse reactions leading to discontinuation of LEXIVA (incidence less than or equal to 1% of subjects) included diarrhea, nausea, vomiting, AST increased, ALT increased, and rash.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
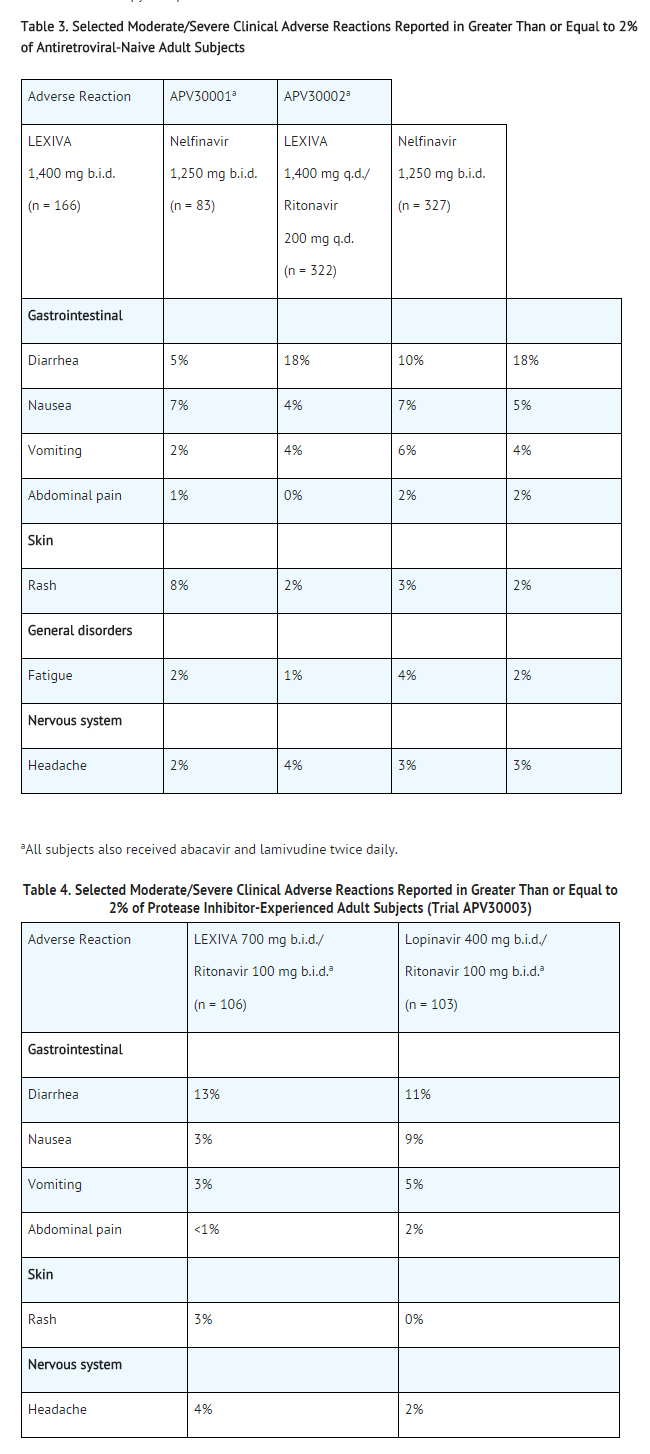
Adult Trials: The data for the 3 active-controlled clinical trials described below reflect exposure of 700 HIV-1–infected subjects to LEXIVA Tablets, including 599 subjects exposed to LEXIVA for greater than 24 weeks, and 409 subjects exposed for greater than 48 weeks. The population age ranged from 17 to 72 years. Of these subjects, 26% were female, 51% Caucasian, 31% black, 16% American Hispanic, and 70% were antiretroviral-naive. Sixty-one percent received LEXIVA 1,400 mg once daily plus ritonavir 200 mg once daily; 24% received LEXIVA 1,400 mg twice daily; and 15% received LEXIVA 700 mg twice daily plus ritonavir 100 mg twice daily.
Selected adverse reactions reported during the clinical efficacy trials of LEXIVA are shown in Tables 3 and 4. Each table presents adverse reactions of moderate or severe intensity in subjects treated with combination therapy for up to 48 weeks.
This image is provided by the National Library of Medicine.
Skin rash (without regard to causality) occurred in approximately 19% of subjects treated with LEXIVA in the pivotal efficacy trials. Rashes were usually maculopapular and of mild or moderate intensity, some with pruritus. Rash had a median onset of 11 days after initiation of LEXIVA and had a median duration of 13 days. Skin rash led to discontinuation of LEXIVA in less than 1% of subjects. In some subjects with mild or moderate rash, dosing with LEXIVA was often continued without interruption; if interrupted, reintroduction of LEXIVA generally did not result in rash recurrence.
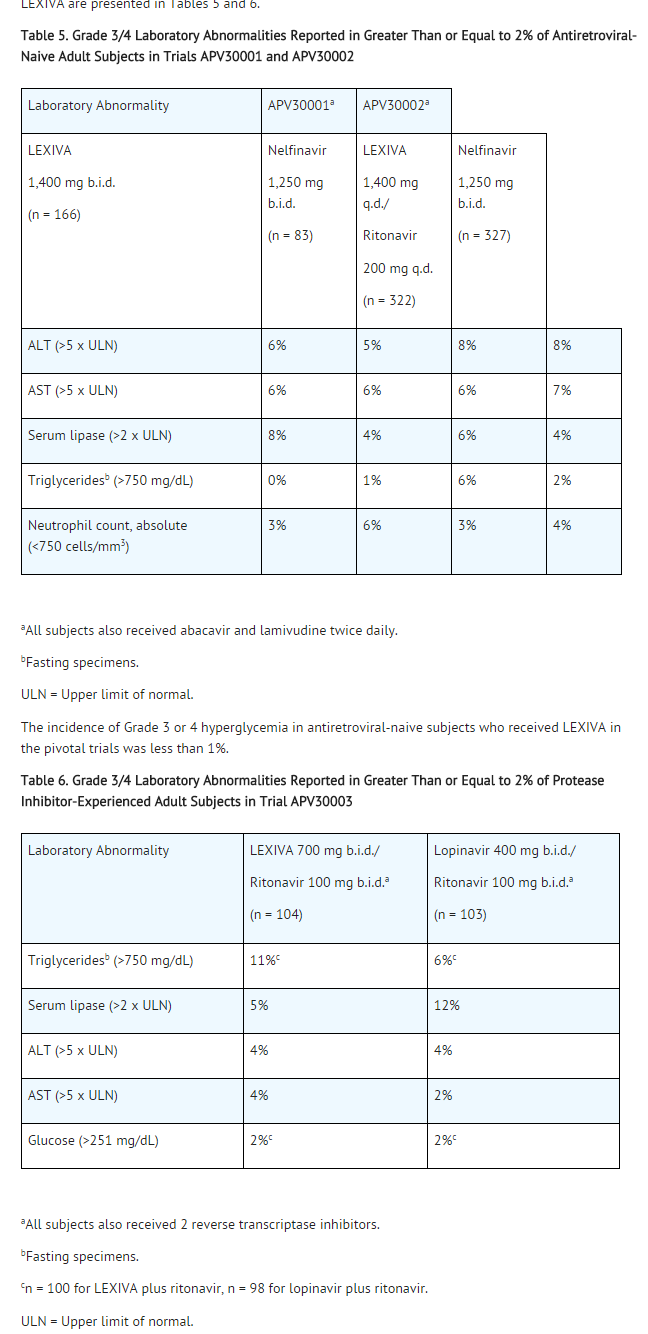
The percentages of subjects with Grade 3 or 4 laboratory abnormalities in the clinical efficacy trials of LEXIVA are presented in Tables 5 and 6.
Table 5. Grade 3/4 Laboratory Abnormalities Reported in Greater Than or Equal to 2% of Antiretroviral-Naive Adult Subjects in Trials APV30001 and APV30002
This image is provided by the National Library of Medicine.
Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following reactions have been identified during post-approval use of LEXIVA. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These reactions have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to LEXIVA.
If LEXIVA is used in combination with ritonavir, see full prescribing information for ritonavir for additional information on drug interactions.
Amprenavir, the active metabolite of fosamprenavir, is an inhibitor of CYP3A4 metabolism and therefore should not be administered concurrently with medications with narrow therapeutic windows that are substrates of CYP3A4. Data also suggest that amprenavir induces CYP3A4.
Amprenavir is metabolized by CYP3A4. Coadministration of LEXIVA and drugs that induce CYP3A4, such as rifampin, may decrease amprenavir concentrations and reduce its therapeutic effect. Coadministration of LEXIVA and drugs that inhibit CYP3A4 may increase amprenavir concentrations and increase the incidence of adverse effects.
The potential for drug interactions with LEXIVA changes when LEXIVA is coadministered with the potent CYP3A4 inhibitor ritonavir. The magnitude of CYP3A4-mediated drug interactions (effect on amprenavir or effect on coadministered drug) may change when LEXIVA is coadministered with ritonavir. Because ritonavir is a CYP2D6 inhibitor, clinically significant interactions with drugs metabolized by CYP2D6 are possible when coadministered with LEXIVA plus ritonavir.
There are other agents that may result in serious and/or life-threatening drug interactions
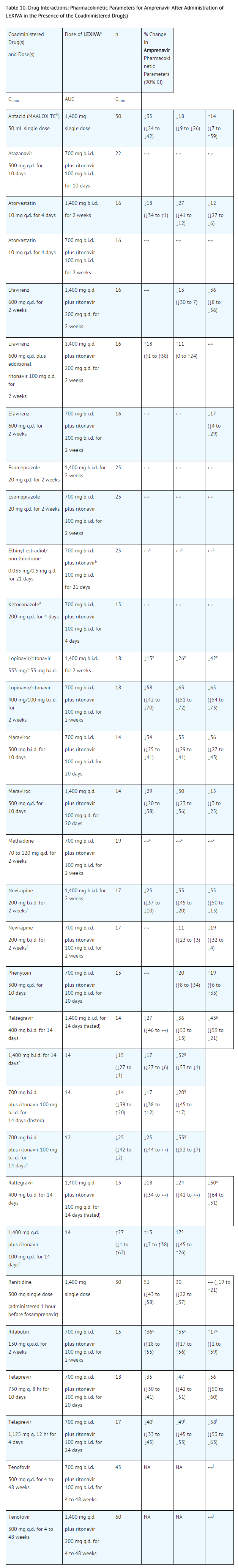
Table 7 provides a listing of established or potentially clinically significant drug interactions. Information in the table applies to LEXIVA with or without ritonavir, unless otherwise indicated.
This image is provided by the National Library of Medicine.
Embryo/fetal development studies were conducted in rats (dosed from day 6 to day 17 of gestation) and rabbits (dosed from day 7 to day 20 of gestation). Administration of fosamprenavir to pregnant rats and rabbits produced no major effects on embryo-fetal development; however, the incidence of abortion was increased in rabbits that were administered fosamprenavir. Systemic exposures (AUC0-24 hr) to amprenavir at these dosages were 0.8 (rabbits) to 2 (rats) times the exposures in humans following administration of the maximum recommended human dose (MRHD) of fosamprenavir alone or 0.3 (rabbits) to 0.7 (rats) times the exposures in humans following administration of the MRHD of fosamprenavir in combination with ritonavir. In contrast, administration of amprenavir was associated with abortions and an increased incidence of minor skeletal variations resulting from deficient ossification of the femur, humerus, and trochlea, in pregnant rabbits at the tested dose approximately one-twentieth the exposure seen at the recommended human dose.
The mating and fertility of the F1 generation born to female rats given fosamprenavir was not different from control animals; however, fosamprenavir did cause a reduction in both pup survival and body weights. Surviving F1 female rats showed an increased time to successful mating, an increased length of gestation, a reduced number of uterine implantation sites per litter, and reduced gestational body weights compared with control animals. Systemic exposure (AUC0-24 hr) to amprenavir in the F0 pregnant rats was approximately 2 times higher than exposures in humans following administration of the MRHD of fosamprenavir alone or approximately the same as those seen in humans following administration of the MRHD of fosamprenavir in combination with ritonavir.
There are no adequate and well-controlled studies in pregnant women. LEXIVA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Antiretroviral Pregnancy Registry: To monitor maternal-fetal outcomes of pregnant women exposed to LEXIVA, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV. Although it is not known if amprenavir is excreted in human milk, amprenavir is secreted into the milk of lactating rats. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breastfeed if they are receiving LEXIVA.
The safety, pharmacokinetic profile, virologic, and immunologic responses of LEXIVA with and without ritonavir were evaluated in protease inhibitor-naive and –experienced HIV-1–infected pediatric subjects aged at least 4 weeks to less than 18 years and weighing at least 3 kg in 3 open-label trials . Vomiting and neutropenia, were more frequent in pediatrics than in adults. Other adverse events occurred with similar frequency in pediatric subjects compared with adults.
Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Fosamprenavir in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Fosamprenavir during labor and delivery.
Nursing Mothers
There is no FDA guidance on the use of Fosamprenavir with respect to nursing mothers.
Pediatric Use
Treatment with LEXIVA is not recommended in protease inhibitor-experienced pediatric patients younger than 6 months. The pharmacokinetics, safety, tolerability, and efficacy of LEXIVA in pediatric patients younger than 4 weeks have not been established. Available pharmacokinetic and clinical data do not support once-daily dosing of LEXIVA alone or in combination with ritonavir for any pediatrics or twice-daily dosing without ritonavir in pediatric patients younger than 2 years.
Geriatic Use
Clinical studies of LEXIVA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger adults. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Fosamprenavir with respect to specific gender populations.
Race
There is no FDA guidance on the use of Fosamprenavir with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Fosamprenavir in patients with renal impairment.
Hepatic Impairment
Amprenavir is principally metabolized by the liver; therefore, caution should be exercised when administering LEXIVA to patients with hepatic impairment because amprenavir concentrations may be increased. Patients with impaired hepatic function receiving LEXIVA with or without concurrent ritonavir require dose reduction.
There are no data to support dosing recommendations for pediatric subjects with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Fosamprenavir in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Fosamprenavir in patients who are immunocompromised.
Monitor cholesterol and triglycerides prior to therapy and periodically thereafter
IV Compatibility
There is limited information regarding IV Compatibility of Fosamprenavir in the drug label.
Overdosage
In a healthy volunteer repeat-dose pharmacokinetic trial evaluating high-dose combinations of LEXIVA plus ritonavir, an increased frequency of Grade 2/3 ALT elevations (greater than 2.5 x ULN) was observed with LEXIVA 1,400 mg twice daily plus ritonavir 200 mg twice daily (4 of 25 subjects). Concurrent Grade 1/2 elevations in AST (greater than 1.25 x ULN) were noted in 3 of these 4 subjects. These transaminase elevations resolved following discontinuation of dosing.
There is no known antidote for LEXIVA. It is not known whether amprenavir can be removed by peritoneal dialysis or hemodialysis. If overdosage occurs, the patient should be monitored for evidence of toxicity and standard supportive treatment applied as necessary.
Pharmacology
There is limited information regarding Fosamprenavir Pharmacology in the drug label.
Mechanism of Action
Fosamprenavir is a prodrug that is rapidly hydrolyzed to amprenavir by cellular phosphatases in the gut epithelium as it is absorbed. Amprenavir is an inhibitor of HIV-1 protease. Amprenavir binds to the active site of HIV-1 protease and thereby prevents the processing of viral Gag and Gag-Pol polyprotein precursors, resulting in the formation of immature non-infectious viral particles.
Antiviral Activity
Fosamprenavir has little or no antiviral activity in cell culture. The antiviral activity of amprenavir was evaluated against HIV-1 IIIB in both acutely and chronically infected lymphoblastic cell lines (MT-4, CEM-CCRF, H9) and in peripheral blood lymphocytes in cell culture. The 50% effective concentration (EC50) of amprenavir ranged from 0.012 to 0.08 microM in acutely infected cells and was 0.41 microM in chronically infected cells (1 microM = 0.50 mcg per mL). The median EC50 value of amprenavir against HIV-1 isolates from clades A to G was 0.00095 microM in peripheral blood mononuclear cells (PBMCs). Similarly, the EC50 values for amprenavir against monocytes/macrophage tropic HIV-1 isolates (clade B) ranged from 0.003 to 0.075 microM in monocyte/macrophage cultures. The EC50 values of amprenavir against HIV-2 isolates grown in PBMCs were higher than those for HIV-1 isolates, and ranged from 0.003 to 0.11 microM. Amprenavir exhibited synergistic anti–HIV–1 activity in combination with the nucleoside reverse transcriptase inhibitors (NRTIs) abacavir, didanosine, lamivudine, stavudine, tenofovir, and zidovudine; the non-nucleoside reverse transcriptase inhibitors (NNRTIs) delavirdine and efavirenz; and the protease inhibitors atazanavir and saquinavir. Amprenavir exhibited additive anti–HIV–1 activity in combination with the NNRTI nevirapine, the protease inhibitors indinavir, lopinavir, nelfinavir, and ritonavir; and the fusion inhibitor enfuvirtide. These drug combinations have not been adequately studied in humans.
Resistance
HIV-1 isolates with decreased susceptibility to amprenavir have been selected in cell culture and obtained from subjects treated with fosamprenavir. Genotypic analysis of isolates from treatment-naive subjects failing amprenavir-containing regimens showed substitutions in the HIV-1 protease gene resulting in amino acid substitutions primarily at positions V32I, M46I/L, I47V, I50V, I54L/M, and I84V, as well as substitutions in the p7/p1 and p1/p6 Gag and Gag-Pol polyprotein precursor cleavage sites. Some of these amprenavir resistance-associated substitutions have also been detected in HIV-1 isolates from antiretroviral-naive subjects treated with LEXIVA. Of the 488 antiretroviral-naive subjects treated with LEXIVA 1,400 mg twice daily or LEXIVA 1,400 mg plus ritonavir 200 mg once daily in Trials APV30001 and APV30002, respectively, 61 subjects (29 receiving LEXIVA and 32 receiving LEXIVA/ritonavir) with virologic failure (plasma HIV-1 RNA greater than 1,000 copies per mL on 2 occasions on or after Week 12) were genotyped. Five of the 29 antiretroviral-naive subjects (17%) receiving LEXIVA without ritonavir in Trial APV30001 had evidence of genotypic resistance to amprenavir: I54L/M (n = 2), I54L + L33F (n = 1), V32I + I47V (n = 1), and M46I + I47V (n = 1). No amprenavir resistance-associated substitutions were detected in antiretroviral-naive subjects treated with LEXIVA/ritonavir for 48 weeks in Trial APV30002. However, the M46I and I50V substitutions were detected in isolates from 1 virologic failure subject receiving LEXIVA/ritonavir once daily at Week 160 (HIV-1 RNA greater than 500 copies per mL). Upon retrospective analysis of stored samples using an ultrasensitive assay, these resistant substitutions were traced back to Week 84 (76 weeks prior to clinical virologic failure).
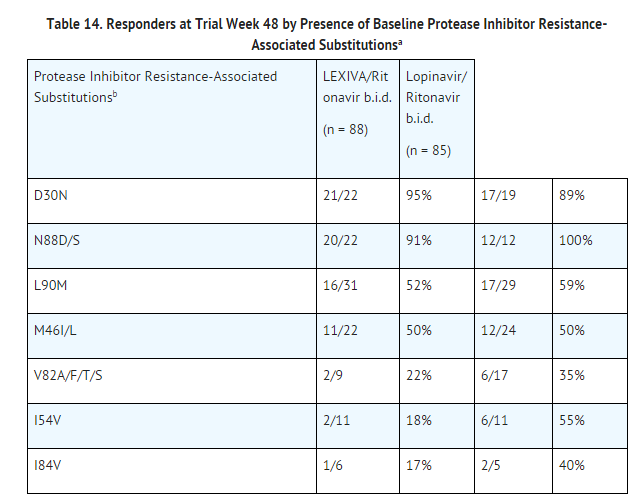
Cross-Resistance: Varying degrees of cross-resistance among HIV-1 protease inhibitors have been observed. An association between virologic response at 48 weeks (HIV-1 RNA level less than 400 copies per mL) and protease inhibitor-resistance substitutions detected in baseline HIV-1 isolates from protease inhibitor-experienced subjects receiving LEXIVA/ritonavir twice daily (n = 88), or lopinavir/ritonavir twice daily (n = 85) in Trial APV30003 is shown in Table 14. The majority of subjects had previously received either one (47%) or 2 protease inhibitors (36%), most commonly nelfinavir (57%) and indinavir (53%). Out of 102 subjects with baseline phenotypes receiving twice-daily LEXIVA/ritonavir, 54% (n = 55) had resistance to at least one protease inhibitor, with 98% (n = 54) of those having resistance to nelfinavir. Out of 97 subjects with baseline phenotypes in the lopinavir/ritonavir arm, 60% (n = 58) had resistance to at least one protease inhibitor, with 97% (n = 56) of those having resistance to nelfinavir.
The virologic response based upon baseline phenotype was assessed. Baseline isolates from protease inhibitor-experienced subjects responding to LEXIVA/ritonavir twice daily had a median shift in susceptibility to amprenavir relative to a standard wild-type reference strain of 0.7 (range: 0.1 to 5.4, n = 62), and baseline isolates from individuals failing therapy had a median shift in susceptibility of 1.9 (range: 0.2 to 14, n = 29). Because this was a select patient population, these data do not constitute definitive clinical susceptibility break points. Additional data are needed to determine clinically relevant break points for LEXIVA.
Isolates from 15 of the 20 subjects receiving twice-daily LEXIVA/ritonavir up to Week 48 and experiencing virologic failure/ongoing replication were subjected to genotypic analysis. The following amprenavir resistance-associated substitutions were found either alone or in combination: V32I, M46I/L, I47V, I50V, I54L/M, and I84V. Isolates from 4 of the 16 subjects continuing to receive twice-daily LEXIVA/ritonavir up to Week 96 who experienced virologic failure underwent genotypic analysis. Isolates from 2 subjects contained amprenavir resistance-associated substitutions: V32I, M46I, and I47V in 1 isolate and I84V in the other.
Structure
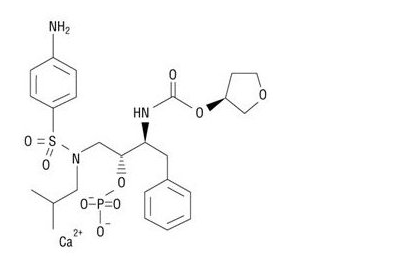
LEXIVA (fosamprenavir calcium) is a prodrug of amprenavir, an inhibitor of HIV protease. The chemical name of fosamprenavir calcium is (3S)-tetrahydrofuran-3-yl (1S,2R)-3-(4-aminophenyl) sulfonyl](isobutyl)amino]-1-benzyl-2-(phosphonooxy) propylcarbamate monocalcium salt. Fosamprenavir calcium is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H34CaN3O9PS and a molecular weight of 623.7. It has the following structural formula:
This image is provided by the National Library of Medicine.
Fosamprenavir calcium is a white to cream-colored solid with a solubility of approximately 0.31 mg per mL in water at 25°C.
LEXIVA Tablets are available for oral administration in a strength of 700 mg of fosamprenavir as fosamprenavir calcium (equivalent to approximately 600 mg of amprenavir). Each 700 mg tablet contains the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and povidone K30. The tablet film-coating contains the inactive ingredients hypromellose, iron oxide red, titanium dioxide, and triacetin.
LEXIVA Oral Suspension is available in a strength of 50 mg per mL of fosamprenavir as fosamprenavir calcium equivalent to approximately 43 mg of amprenavir. * LEXIVA Oral Suspension is a white to off-white suspension with a grape-bubblegum-peppermint flavor. Each one milliliter (1 mL) contains the inactive ingredients artificial grape-bubblegum flavor, calcium chloride dihydrate, hypromellose, methylparaben, natural peppermint flavor, polysorbate 80, propylene glycol, propylparaben, purified water, and sucralose.
Pharmacodynamics
There is limited information regarding Pharmacodynamics of Fosamprenavir in the drug label.
Pharmacokinetics
The pharmacokinetic properties of amprenavir after administration of LEXIVA, with or without ritonavir, have been evaluated in both healthy adult volunteers and in HIV-1-infected subjects; no substantial differences in steady-state amprenavir concentrations were observed between the 2 populations.
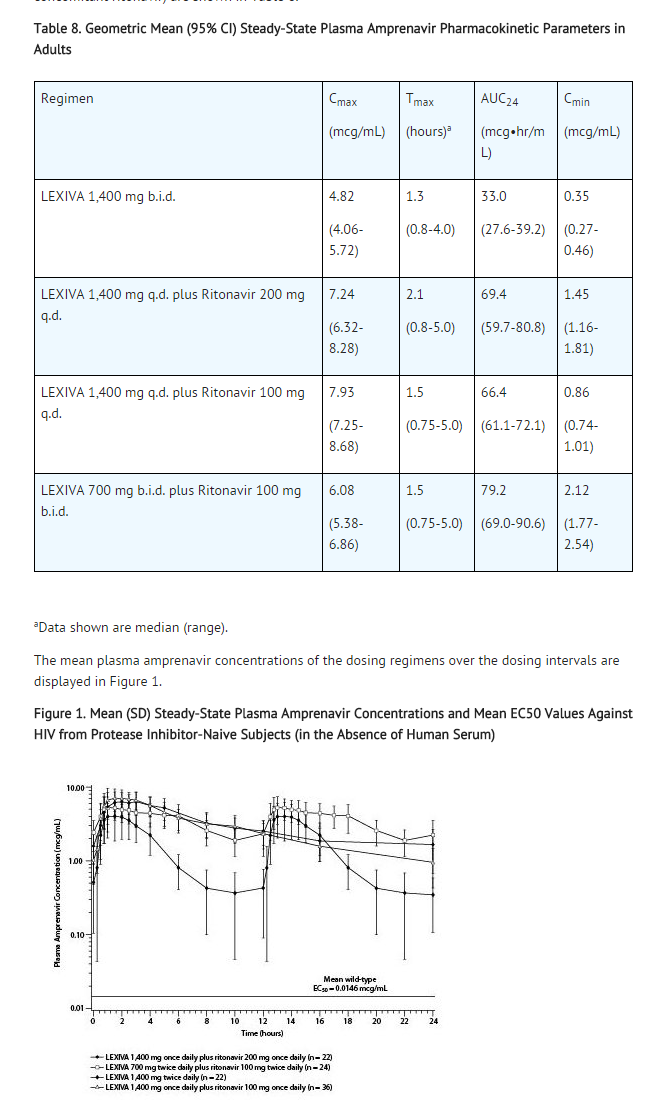
The pharmacokinetic parameters of amprenavir after administration of LEXIVA (with and without concomitant ritonavir) are shown in Table 8.
This image is provided by the National Library of Medicine.
Absorption and Bioavailability
After administration of a single dose of LEXIVA to HIV-1–infected subjects, the time to peak amprenavir concentration (Tmax) occurred between 1.5 and 4 hours (median 2.5 hours). The absolute oral bioavailability of amprenavir after administration of LEXIVA in humans has not been established.
After administration of a single 1,400-mg dose in the fasted state, LEXIVA Oral Suspension (50 mg per mL) and LEXIVA Tablets (700 mg) provided similar amprenavir exposures (AUC); however, the Cmax of amprenavir after administration of the suspension formulation was 14.5% higher compared with the tablet.
Effects of Food on Oral Absorption: Administration of a single 1,400-mg dose of LEXIVA Tablets in the fed state (standardized high-fat meal: 967 kcal, 67 grams fat, 33 grams protein, 58 grams carbohydrate) compared with the fasted state was associated with no significant changes in amprenavir Cmax, Tmax, or AUC.
Administration of a single 1,400-mg dose of LEXIVA Oral Suspension in the fed state (standardized high-fat meal: 967 kcal, 67 grams fat, 33 grams protein, 58 grams carbohydrate) compared with the fasted state was associated with a 46% reduction in Cmax, a 0.72-hour delay in Tmax, and a 28% reduction in amprenavir AUC0.
Distribution
In vitro, amprenavir is approximately 90% bound to plasma proteins, primarily to alpha1-acid glycoprotein. In vitro, concentration-dependent binding was observed over the concentration range of 1 to 10 mcg per mL, with decreased binding at higher concentrations. The partitioning of amprenavir into erythrocytes is low, but increases as amprenavir concentrations increase, reflecting the higher amount of unbound drug at higher concentrations.
Metabolism
After oral administration, fosamprenavir is rapidly and almost completely hydrolyzed to amprenavir and inorganic phosphate prior to reaching the systemic circulation. This occurs in the gut epithelium during absorption. Amprenavir is metabolized in the liver by the CYP3A4 enzyme system. The 2 major metabolites result from oxidation of the tetrahydrofuran and aniline moieties. Glucuronide conjugates of oxidized metabolites have been identified as minor metabolites in urine and feces.
Amprenavir is both a substrate for and inducer of P-glycoprotein.
Elimination
Excretion of unchanged amprenavir in urine and feces is minimal. Unchanged amprenavir in urine accounts for approximately 1% of the dose; unchanged amprenavir was not detectable in feces. Approximately 14% and 75% of an administered single dose of 14C-amprenavir can be accounted for as metabolites in urine and feces, respectively. Two metabolites accounted for greater than 90% of the radiocarbon in fecal samples. The plasma elimination half-life of amprenavir is approximately 7.7 hours.
Special Populations
Hepatic Impairment: The pharmacokinetics of amprenavir have been studied after the administration of LEXIVA in combination with ritonavir to adult HIV-1–infected subjects with mild, moderate, and severe hepatic impairment. Following 2 weeks of dosing with LEXIVA plus ritonavir, the AUC of amprenavir was increased by approximately 22% in subjects with mild hepatic impairment, by approximately 70% in subjects with moderate hepatic impairment, and by approximately 80% in subjects with severe hepatic impairment compared with HIV-1–infected subjects with normal hepatic function. Protein binding of amprenavir was decreased in subjects with hepatic impairment. The unbound fraction at 2 hours (approximate Cmax) ranged between a decrease of -7% to an increase of 57% while the unbound fraction at the end of the dosing interval (Cmin) increased from 50% to 102%.
The pharmacokinetics of amprenavir have been studied after administration of amprenavir given as AGENERASE® Capsules to adult subjects with hepatic impairment. Following administration of a single 600-mg oral dose, the AUC of amprenavir was increased by approximately 2.5-fold in subjects with moderate cirrhosis and by approximately 4.5-fold in subjects with severe cirrhosis compared with healthy volunteers.
Renal Impairment
The impact of renal impairment on amprenavir elimination in adults has not been studied. The renal elimination of unchanged amprenavir represents approximately 1% of the administered dose; therefore, renal impairment is not expected to significantly impact the elimination of amprenavir.
Pediatric Patients
The pharmacokinetics of amprenavir following administration of LEXIVA Oral Suspension and LEXIVA Tablets, with or without ritonavir, have been studied in a total of 212 HIV-1–infected pediatric subjects enrolled in 3 trials. LEXIVA without ritonavir was administered as 30 or 40 mg per kg twice daily to children aged 2 to 5 years. LEXIVA with ritonavir was administered as LEXIVA 30 mg per kg plus ritonavir 6 mg per kg once daily to children aged 2 to 18 years and as LEXIVA 18 to 60 mg per kg plus ritonavir 3 to 10 mg per kg twice daily to children aged at least 4 weeks to 18 years; body weights ranged from 3 to 103 kg.
Amprenavir apparent clearance decreased with increasing weight. Weight-adjusted apparent clearance was higher in children younger than 4 years, suggesting that younger children require higher mg per kg dosing of LEXIVA.
The pharmacokinetics of LEXIVA Oral Suspension in protease inhibitor-naive infants younger than 6 months (n = 9) receiving LEXIVA 45 mg per kg plus ritonavir 10 mg per kg twice daily generally demonstrated lower AUC12 and Cmin than adults receiving twice-daily LEXIVA 700 mg plus ritonavir 100 mg, the dose recommended for protease-experienced adults. The mean steady-state amprenavir AUC12, Cmax, and Cmin were 26.6 mcg•hour per mL, 6.25 mcg per mL, and 0.86 mcg per mL, respectively. These data do not support twice-daily dosing of LEXIVA alone or in combination with ritonavir in protease inhibitor-experienced patients younger than 6 months. Because of expected low amprenavir exposure and a requirement for large volume of drug, twice-daily dosing of LEXIVA alone (without ritonavir) in pediatric subjects younger than 2 years was not studied.
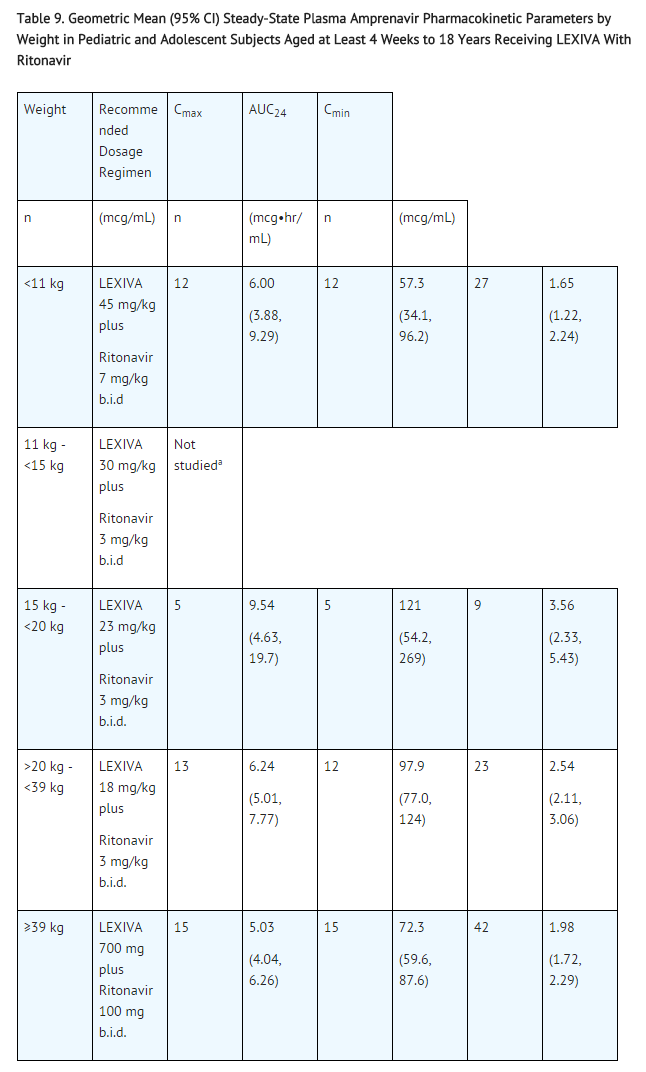
Pharmacokinetic parameters for LEXIVA administered with food and with ritonavir in this patient population at the recommended weight-band–based dosage regimens are provided in Table 9.
This image is provided by the National Library of Medicine.
This image is provided by the National Library of Medicine.
This image is provided by the National Library of Medicine.
This image is provided by the National Library of Medicine.
Nonclinical Toxicology
In long-term carcinogenicity studies, fosamprenavir was administered orally for up to 104 weeks at doses of 250, 400, or 600 mg per kg per day in mice and at doses of 300, 825, or 2,250 mg per kg per day in rats. Exposures at these doses were 0.3- to 0.7-fold (mice) and 0.7- to 1.4-fold (rats) those in humans given 1,400 mg twice daily of fosamprenavir alone, and 0.2- to 0.3-fold (mice) and 0.3- to 0.7-fold (rats) those in humans given 1,400 mg once daily of fosamprenavir plus 200 mg ritonavir once daily. Exposures in the carcinogenicity studies were 0.1- to 0.3-fold (mice) and 0.3- to 0.6-fold (rats) those in humans given 700 mg of fosamprenavir plus 100 mg ritonavir twice daily. There was an increase in hepatocellular adenomas and hepatocellular carcinomas at all doses in male mice and at 600 mg per kg per day in female mice, and in hepatocellular adenomas and thyroid follicular cell adenomas at all doses in male rats, and at 835 mg per kg per day and 2,250 mg per kg per day in female rats. The relevance of the hepatocellular findings in the rodents for humans is uncertain. Repeat dose studies with fosamprenavir in rats produced effects consistent with enzyme induction, which predisposes rats, but not humans, to thyroid neoplasms. In addition, in rats only there was an increase in interstitial cell hyperplasia at 825 mg per kg per day and 2,250 mg per kg per day, and an increase in uterine endometrial adenocarcinoma at 2,250 mg per kg per day. The incidence of endometrial findings was slightly increased over concurrent controls, but was within background range for female rats. The relevance of the uterine endometrial adenocarcinoma findings in rats for humans is uncertain.
Fosamprenavir was not mutagenic or genotoxic in a battery of in vitro and in vivo assays. These assays included bacterial reverse mutation (Ames), mouse lymphoma, rat micronucleus, and chromosome aberrations in human lymphocytes.
The effects of fosamprenavir on fertility and general reproductive performance were investigated in male (treated for 4 weeks before mating) and female rats (treated for 2 weeks before mating through postpartum day 6). Systemic exposures (AUC0-24 hr) to amprenavir in these studies were 3 (males) to 4 (females) times higher than exposures in humans following administration of the MRHD of fosamprenavir alone or similar to those seen in humans following administration of fosamprenavir in combination with ritonavir.
Fosamprenavir did not impair mating or fertility of male or female rats and did not affect the development and maturation of sperm from treated rats.
Clinical Studies
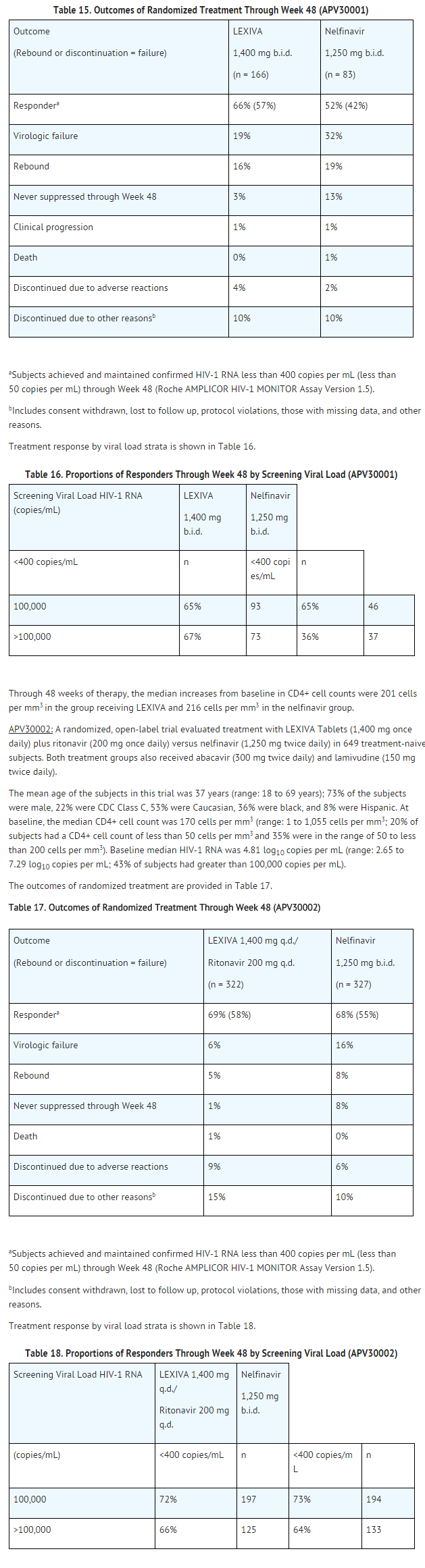
APV30001: A randomized, open-label trial evaluated treatment with LEXIVA Tablets (1,400 mg twice daily) versus nelfinavir (1,250 mg twice daily) in 249 antiretroviral treatment-naive subjects. Both groups of subjects also received abacavir (300 mg twice daily) and lamivudine (150 mg twice daily).
The mean age of the subjects in this trial was 37 years (range: 17 to 70 years); 69% of the subjects were male, 20% were CDC Class C (AIDS), 24% were Caucasian, 32% were black, and 44% were Hispanic. At baseline, the median CD4+ cell count was 212 cells per mm3 (range: 2 to 1,136 cells per mm3; 18% of subjects had a CD4+ cell count of less than 50 cells per mm3 and 30% were in the range of 50 to less than 200 cells per mm3). Baseline median HIV-1 RNA was 4.83 log10 copies per mL (range: 1.69 to 7.41 log10 copies per mL; 45% of subjects had greater than 100,000 copies per mL).
The outcomes of randomized treatment are provided in Table 15.
This image is provided by the National Library of Medicine.
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 203 cells per mm3 in the group receiving LEXIVA and 207 cells per mm3 in the nelfinavir group.
APV30003
A randomized, open-label, multicenter trial evaluated 2 different regimens of LEXIVA plus ritonavir (LEXIVA Tablets 700 mg twice daily plus ritonavir 100 mg twice daily or LEXIVA Tablets 1,400 mg once daily plus ritonavir 200 mg once daily) versus lopinavir/ritonavir (400 mg/100 mg twice daily) in 315 subjects who had experienced virologic failure to 1 or 2 prior protease inhibitor-containing regimens.
The mean age of the subjects in this trial was 42 years (range: 24 to 72 years); 85% were male, 33% were CDC Class C, 67% were Caucasian, 24% were black, and 9% were Hispanic. The median CD4+ cell count at baseline was 263 cells per mm3 (range: 2 to 1,171 cells per mm3). Baseline median plasma HIV-1 RNA level was 4.14 log10 copies per mL (range: 1.69 to 6.41 log10 copies per mL).
The median durations of prior exposure to NRTIs were 257 weeks for subjects receiving LEXIVA/ritonavir twice daily (79% had greater than or equal to 3 prior NRTIs) and 210 weeks for subjects receiving lopinavir/ritonavir (64% had greater than or equal to 3 prior NRTIs). The median durations of prior exposure to protease inhibitors were 149 weeks for subjects receiving LEXIVA/ritonavir twice daily (49% received greater than or equal to 2 prior protease inhibitors) and 130 weeks for subjects receiving lopinavir/ritonavir (40% received greater than or equal to 2 prior protease inhibitors).
The time-averaged changes in plasma HIV-1 RNA from baseline (AAUCMB) at 48 weeks (the endpoint on which the trial was powered) were -1.4 log10 copies per mL for twice-daily LEXIVA/ritonavir and -1.67 log10 copies per mL for the lopinavir/ritonavir group.
The proportions of subjects who achieved and maintained confirmed HIV-1 RNA less than 400 copies per mL (secondary efficacy endpoint) were 58% with twice-daily LEXIVA/ritonavir and 61% with lopinavir/ritonavir (95% CI for the difference: -16.6, 10.1). The proportions of subjects with HIV-1 RNA less than 50 copies per mL with twice-daily LEXIVA/ritonavir and with lopinavir/ritonavir were 46% and 50%, respectively (95% CI for the difference: -18.3, 8.9). The proportions of subjects who were virologic failures were 29% with twice-daily LEXIVA/ritonavir and 27% with lopinavir/ritonavir.
The frequency of discontinuations due to adverse events and other reasons, and deaths were similar between treatment arms.
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 81 cells per mm3 with twice-daily LEXIVA/ritonavir and 91 cells per mm3 with lopinavir/ritonavir.
This trial was not large enough to reach a definitive conclusion that LEXIVA/ritonavir and lopinavir/ritonavir are clinically equivalent.
Once-daily administration of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced patients. Through Week 48, 50% and 37% of subjects receiving LEXIVA 1,400 mg plus ritonavir 200 mg once daily had plasma HIV-1 RNA less than 400 copies per mL and less than 50 copies per mL, respectively.
Three open-label trials in pediatric subjects aged at least 4 weeks to 18 years were conducted. In one trial (APV29005), twice-daily dosing regimens (LEXIVA with or without ritonavir) were evaluated in combination with other antiretroviral agents in pediatric subjects aged 2 to 18 years. In a second trial (APV20002), twice-daily dosing regimens (LEXIVA with ritonavir) were evaluated in combination with other antiretroviral agents in pediatric subjects aged at least 4 weeks to less than 2 years. A third trial (APV20003) evaluated once-daily dosing of LEXIVA with ritonavir; the pharmacokinetic data from this trial did not support a once-daily dosing regimen in any pediatric patient population.
APV29005
LEXIVA: Twenty (18 therapy-naive and 2 therapy-experienced) pediatric subjects received LEXIVA Oral Suspension without ritonavir twice daily. At Week 24, 65% (13/20) achieved HIV-1 RNA less than 400 copies per mL, and the median increase from baseline in CD4+ cell count was 350 cells per mm3.
LEXIVA plus Ritonavir: Forty-nine protease inhibitor-naive and 40 protease inhibitor-experienced pediatric subjects received LEXIVA Oral Suspension or Tablets with ritonavir twice daily. At Week 24, 71% of protease inhibitor-naive (35/49) and 55% of protease inhibitor-experienced (22/40) subjects achieved HIV-1 RNA less than 400 copies per mL; median increases from baseline in CD4+ cell counts were 184 cells per mm3 and 150 cells per mm3 in protease inhibitor-naive and experienced subjects, respectively.
APV20002
Fifty-four pediatric subjects (49 protease inhibitor-naive and 5 protease inhibitor-experienced) received LEXIVA Oral Suspension with ritonavir twice daily. At Week 24, 72% of subjects achieved HIV-1 RNA less than 400 copies per mL. The median increases from baseline in CD4+ cell counts were 400 cells per mm3 in subjects aged at least 4 weeks to less than 6 months and 278 cells per mm3 in subjects aged 6 months to 2 years.
How Supplied
LEXIVA Tablets, 700 mg, are pink, film-coated, capsule-shaped, biconvex tablets, with “GX LL7” debossed on one face.
Bottle of 60 with child-resistant closure (NDC 49702-207-18).
Store at controlled room temperature of 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) (see USP Controlled Room Temperature). Keep container tightly closed.
LEXIVA Oral Suspension, a white to off-white grape-bubblegum-peppermint–flavored suspension, contains 50 mg of fosamprenavir as fosamprenavir calcium equivalent to approximately 43 mg of amprenavir in each 1 mL.
Bottle of 225 mL with child-resistant closure (NDC 49702-208-53).
This product does not require reconstitution.
Storage
Store in refrigerator or at room temperature (5° to 30°C; 41° to 86°F). Shake vigorously before using. Do not freeze.
Images
Drug Images
Package and Label Display Panel
This image is provided by the National Library of Medicine.This image is provided by the National Library of Medicine.This image is provided by the National Library of Medicine.
This image of the FDA label is provided by the National Library of Medicine.
This image of the FDA label is provided by the National Library of Medicine.
This image of the FDA label is provided by the National Library of Medicine.
Patient Counseling Information
A statement to patients and healthcare providers is included on the product’s bottle label: ALERT: Find out about medicines that should NOT be taken with LEXIVA.
LEXIVA may interact with many drugs; therefore, patients should be advised to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, particularly St. John’s wort.
Patients receiving PDE5 inhibitors should be advised that they may be at an increased risk of PDE5 inhibitor-associated adverse events, including hypotension, visual changes, and priapism, and should promptly report any symptoms to their healthcare provider.
Patients receiving hormonal contraceptives should be instructed to use alternate contraceptive measures during therapy with LEXIVA because hormonal levels may be altered, and if used in combination with LEXIVA and ritonavir, liver enzyme elevations may occur.
Patients should inform their healthcare provider if they have a sulfa allergy. The potential for cross-sensitivity between drugs in the sulfonamide class and fosamprenavir is unknown.
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy, including LEXIVA, and that the cause and long-term health effects of these conditions are not known at this time.
LEXIVA is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using LEXIVA.
Patients should be advised to avoid doing things that can spread HIV-1 infection to others.
Do not share needles or other injection equipment.
Do not share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
Do not have any kind of sex without protection.
Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.•
Do not breastfeed.
We do not know if LEXIVA can be passed to your baby in your breast milk and whether it could harm your baby. Also, mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.
Patients should be told that sustained decreases in plasma HIV-1 RNA have been associated with a reduced risk of progression to AIDS and death.
Patients should be advised to take LEXIVA every day as prescribed. LEXIVA must always be used in combination with other antiretroviral drugs. Patients should not alter the dose or discontinue therapy without consulting their physician. If a dose is missed, patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped, the patient should not double the next dose.
Patients should be instructed to shake the bottle vigorously before each use and that refrigeration of the oral suspension may improve the taste for some patients.
LEXIVA and AGENERASE are registered trademarks of ViiV Healthcare.
The brands listed are trademarks of their respective owners and are not trademarks of ViiV Healthcare. The makers of these brands are not affiliated with and do not endorse ViiV Healthcare or its products.
Precautions with Alcohol
Alcohol-Fosamprenavir interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.














{kind=link}


