Darbepoetin alfa
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
WARNING: ESAS INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE
See full prescribing information for complete Boxed Warning.
Chronic Kidney Disease:
Cancer
|
Overview
Darbepoetin alfa is a colony stimulating factor that is FDA approved for the treatment of anemia. There is a Black Box Warning for this drug as shown here. Common adverse reactions include Seizures, PRCA, allergy.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Anemia Due to Chronic Kidney Disease
- Darbepoetin alfa is indicated for the treatment of anemia due to chronic kidney disease (CKD), including patients on dialysis and patients not on dialysis.
Anemia Due to Chemotherapy in Patients With Cancer
- Darbepoetin alfa is indicated for the treatment of anemia in patients with non-myeloid malignancies where anemia is due to the effect of concomitant myelosuppressive chemotherapy, and upon initiation, there is a minimum of two additional months of planned chemotherapy.
Limitations of Use
- Darbepoetin alfa has not been shown to improve quality of life, fatigue, or patient well-being.
- Darbepoetin alfa is not indicated for use:
- In patients with cancer receiving hormonal agents, biologic products, or radiotherapy, unless also receiving concomitant myelosuppressive chemotherapy.
- In patients with cancer receiving myelosuppressive chemotherapy when the anticipated outcome is cure.
- As a substitute for RBC transfusions in patients who require immediate correction of anemia.
Evaluation of Iron Stores and Nutritional Factors
- Evaluate the iron status in all patients before and during treatment and maintain iron repletion. Correct or exclude other causes of anemia (e.g., vitamin deficiency, metabolic or chronic inflammatory conditions,bleeding, etc.) before initiating darbepoetin alfa.
Patients with Chronic Kidney Disease
In controlled trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered erythropoiesis-stimulating agents (ESAs) to target a hemoglobin level of greater than 11 g/dL. No trial has identified a hemoglobin target level, darbepoetin alfa dose, or dosing strategy that does not increase these risks. Individualize dosing and use the lowest dose of darbepoetin alfa sufficient to reduce the need for RBC transfusions.
- Physicians and patients should weigh the possible benefits of decreasing transfusions against the increased risks of death and other serious cardiovascular adverse events.
- For all patients with CKD.
- When initiating or adjusting therapy, monitor hemoglobin levels at least weekly until stable, then monitor at least monthly. When adjusting therapy consider hemoglobin rate of rise, rate of decline, ESA responsiveness and hemoglobin variability. A single hemoglobin excursion may not require a dosing change.
- Do not increase the dose more frequently than once every 4 weeks. Decreases in dose can occur more frequently. Avoid frequent dose adjustments.
- If the hemoglobin rises rapidly (e.g., more than 1 g/dL in any 2-week period), reduce the dose of darbepoetin alfa by 25% or more as needed to reduce rapid responses.
- For patients who do not respond adequately, if the hemoglobin has not increased by more than 1 g/dL after 4 weeks of therapy, increase the dose by 25%.
- For patients who do not respond adequately over a 12-week escalation period, increasing the darbepoetin alfa dose further is unlikely to improve response and may increase risks. Use the lowest dose that will maintain a hemoglobin level sufficient to reduce the need for RBC transfusions. Evaluate other causes of anemia. Discontinue darbepoetin alfa if responsiveness does not improve.
- For patients with CKD on dialysis:
- Initiate darbepoetin alfa treatment when the hemoglobin level is less than 10 g/dL.
- If the hemoglobin level approaches or exceeds 11 g/dL, reduce or interrupt the dose of darbepoetin alfa.
- The recommended starting dose is 0.45 mcg/kg intravenously or subcutaneously as a weekly injection or 0.75 mcg/kg once every 2 weeks as appropriate. The intravenous route is recommended for patients on hemodialysis.
- Consider initiating darbepoetin alfa treatment only when the hemoglobin level is less than 10 g/dL and the following considerations apply:
- The rate of hemoglobin decline indicates the likelihood of requiring a RBC transfusion and,
- Reducing the risk of alloimmunization and/or other RBC transfusion-related risks is a goal.
- If the hemoglobin level exceeds 10 g/dL, reduce or interrupt the dose of darbepoetin alfa, and use the lowest dose of darbepoetin alfa sufficient to reduce the need for RBC transfusions.
- The recommended starting dose is 0.45 mcg/kg body weight intravenously or subcutaneously given once at four week intervals as appropriate.
- Conversion from Epoetin alfa to darbepoetin alfa in patients with CKD on dialysis.
- Darbepoetin alfa is administered less frequently than epoetin alfa.
- Administer darbepoetin alfa once weekly in patients who were receiving epoetin alfa 2 to 3 times weekly.
- Administer darbepoetin alfa once every 2 weeks in patients who were receiving epoetin alfa once weekly.
- Estimate the starting weekly dose of darbepoetin alfa for adults and pediatric patients on the basis of the weekly epoetin alfa dose at the time of substitution (see Table 1). Maintain the route of administration (intravenous or subcutaneous injection).

This image is provided by the National Library of Medicine.

- Conversion from Epoetin alfa to darbepoetin alfa in patients with CKD not on dialysis
- The dose conversion depicted in Table 1 does not accurately estimate the once monthly dose of darbepoetin alfa.
Patients on Cancer Chemotherapy
- Initiate darbepoetin alfa in patients on cancer chemotherapy only if the hemoglobin is less than 10 g/dL, and if there is a minimum of two additional months of planned chemotherapy.
- Use the lowest dose of darbepoetin alfa necessary to avoid RBC transfusions.
Recommended Starting Dose
- The recommended starting dose and schedules are:
- 2.25 mcg/kg every week subcutaneously until completion of a chemotherapy course
- 500 mcg every 3 weeks subcutaneously until completion of a chemotherapy course

This image is provided by the National Library of Medicine.

Preparation and Administration
- The needle cover of the prefilled syringe contains dry natural rubber (a derivative of latex), which may cause allergic reactions.
- Do not shake. Do not use darbepoetin alfa that has been shaken or frozen.
- Protect vials and prefilled syringes from light.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use any vials or prefilled syringes exhibiting particulate matter or discoloration.
- Discard unused portion of darbepoetin alfa in vials or prefilled syringes. Do not re-enter vial.
- Do not dilute darbepoetin alfa and do not administer in conjunction with other drug solutions.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Darbepoetin alfa in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Darbepoetin alfa in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
There is limited information regarding FDA-Labeled Use of Darbepoetin alfa in pediatric patients.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Darbepoetin alfa in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Darbepoetin alfa in pediatric patients.
Contraindications
- Darbepoetin alfa is contraindicated in patients with:
- Uncontrolled hypertension
- Pure red cell aplasia (PRCA) that begins after treatment with darbepoetin alfa or other erythropoietin protein drugs
- Serious allergic reactions to darbepoetin alfa
Warnings
|
WARNING: ESAS INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE
See full prescribing information for complete Boxed Warning.
Chronic Kidney Disease:
Cancer
|
Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism
- In controlled clinical trials of patients with CKD comparing higher hemoglobin targets (13 - 14 g/dL) to lower targets (9 - 11.3 g/dL), darbepoetin alfa and other ESAs increased the risk of death, myocardial infarction, stroke, congestive heart failure, thrombosis of hemodialysis vascular access, and other thromboembolic events in the higher target groups.
- Using darbepoetin alfa to target a hemoglobin level of greater than 11 g/dL increases the risk of serious adverse cardiovascular reactions and has not been shown to provide additional benefit.
- Use caution in patients with coexistent cardiovascular disease and stroke.
- Patients with CKD and an insufficient hemoglobin response to ESA therapy may be at even greater risk for cardiovascular reactions and mortality than other patients. A rate of hemoglobin rise of greater than 1 g/dL over 2 weeks may contribute to these risks.
- In controlled clinical trials of patients with cancer, darbepoetin alfa and other ESAs increased the risks for death and serious adverse cardiovascular reactions. These adverse reactions included myocardial infarction and stroke.
- In controlled clinical trials, ESAs increased the risk of death in patients undergoing coronary artery bypass graft surgery (CABG) and the risk of deep venous thrombosis (DVT) in patients undergoing orthopedic procedures.
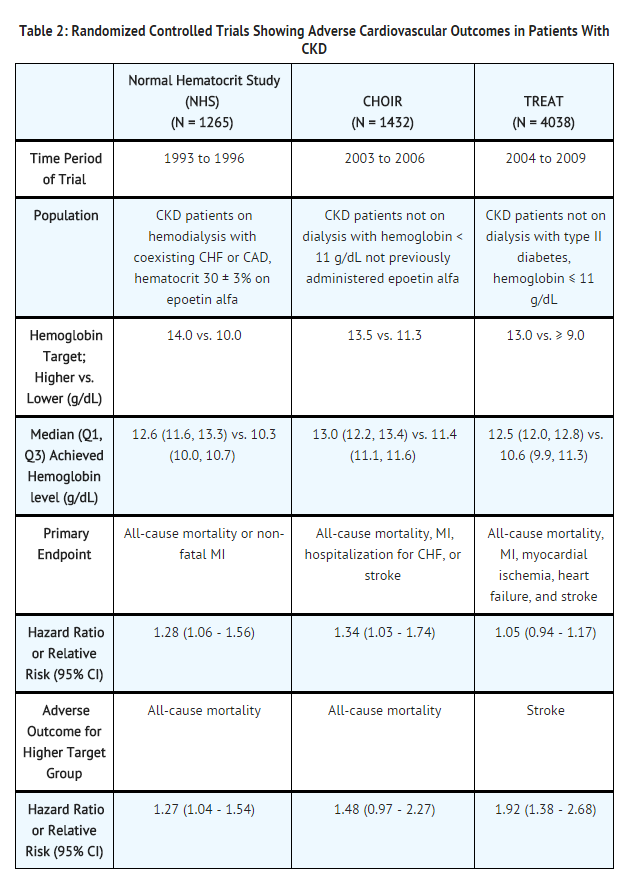
- The design and overall results of the 3 large trials comparing higher and lower hemoglobin targets are shown in Table 2.
This image is provided by the National Library of Medicine.

Patients with Chronic Kidney Disease
- Normal Hematocrit Study (NHS)
- A prospective, randomized, open-label study of 1265 patients with chronic kidney disease on dialysis with documented evidence of congestive heart failure or ischemic heart disease was designed to test the hypothesis that a higher target hematocrit (Hct) would result in improved outcomes compared with a lower target Hct. In this study, patients were randomized to epoetin alfa treatment targeted to a maintenance hemoglobin of either 14 ± 1 g/dL or 10 ± 1 g/dL. The trial was terminated early with adverse safety findings of higher mortality in the high hematocrit target group.
- Higher mortality (35% vs. 29%) was observed for the patients randomized to a target hemoglobin of 14 g/dL than for the patients randomized to a target hemoglobin of 10 g/dL. For all-cause mortality, the HR = 1.27; 95% CI (1.04, 1.54); p=0.018. The incidence of nonfatal myocardial infarction, vascular access thrombosis, and other thrombotic events was also higher in the group randomized to a target hemoglobin of 14 g/dL.
- CHOIR
- A randomized, prospective trial, 1432 patients with anemia due to CKD who were not undergoing dialysis and who had not previously received epoetin alfa therapy were randomized to epoetin alfa treatment targeting a maintenance hemoglobin concentration of either 13.5 g/dL or 11.3 g/dL. The trial was terminated early with adverse safety findings. A major cardiovascular event (death, myocardial infarction, stroke, or hospitalization for congestive heart failure) occurred in 125 of the 715 patients (18%) in the higher hemoglobin group compared to 97 of the 717 patients (14%) in the lower hemoglobin group [hazard ratio (HR) 1.34, 95% CI: 1.03, 1.74; p = 0.03].
- TREAT
- A randomized, double-blind, placebo-controlled, prospective trial of 4038 patients with CKD not on dialysis (eGFR of 20 – 60 mL/min), anemia (hemoglobin levels ≤ 11 g/dL), and type 2 diabetes mellitus, patients were randomized to receive either darbepoetin alfa treatment or a matching placebo. Placebo group patients also received darbepoetin alfa when their hemoglobin levels were below 9 g/dL. The trial objectives were to demonstrate the benefit of darbepoetin alfa treatment of the anemia to a target hemoglobin level of 13 g/dL, when compared to a "placebo" group, by reducing the occurrence of either of two primary endpoints: (1) a composite cardiovascular endpoint of all-cause mortality or a specified cardiovascular event (myocardial ischemia, CHF, MI, and CVA) or (2) a composite renal endpoint of all-cause mortality or progression to end stage renal disease. The overall risks for each of the two primary endpoints (the cardiovascular composite and the renal composite) were not reduced with darbepoetin alfa treatment (see Table 2), but the risk of stroke was increased nearly two-fold in the darbepoetin alfa-treated group versus the placebo group: annualized stroke rate 2.1% vs. 1.1%, respectively, HR 1.92; 95% CI: 1.38, 2.68; p < 0.001. The relative risk of stroke was particularly high in patients with a prior stroke: annualized stroke rate 5.2% in the darbepoetin alfa treated group and 1.9% in the placebo group, HR 3.07; 95% CI: 1.44, 6.54. Also, among darbepoetin alfa-treated subjects with a past history of cancer, there were more deaths due to all causes and more deaths adjudicated as due to cancer, in comparison with the control group.
Patients with Cancer
- An increased incidence of thromboembolic reactions, some serious and life-threatening, occurred in patients with cancer treated with ESAs.
- In a randomized, placebo-controlled study (Study 1 in Table 3 of 939 women with metastatic breast cancer receiving chemotherapy, patients received either weekly epoetin alfa or placebo for up to a year. This study was designed to show that survival was superior when epoetin alfa was administered to prevent anemia (maintain hemoglobin levels between 12 and 14 g/dL or hematocrit between 36% and 42%). This study was terminated prematurely when interim results demonstrated a higher mortality at 4 months (8.7% vs. 3.4%) and a higher rate of fatal thrombotic reactions (1.1% vs. 0.2%) in the first 4 months of the study among patients treated with epoetin alfa. Based on Kaplan-Meier estimates, at the time of study termination, the 12-month survival was lower in the epoetin alfa group than in the placebo group (70% vs. 76%; HR 1.37, 95% CI: 1.07, 1.75; p = 0.012).
Patients Having Surgery
- Darbepoetin alfa is not approved for reduction of RBC transfusions in patients scheduled for surgical procedures.
- An increased incidence of DVT in patients receiving epoetin alfa undergoing surgical orthopedic procedures was demonstrated. In a randomized, controlled study, 680 adult patients, not receiving prophylactic anticoagulation and undergoing spinal surgery, received epoetin alfa and standard of care (SOC) treatment (n = 340) or SOC treatment alone (n = 340). A higher incidence of DVTs, determined by either color flow duplex imaging or by clinical symptoms, was observed in the epoetin alfa group (16 [4.7%] patients) compared with the SOC group (7 [2.1%] patients). In addition to the 23 patients with DVTs included in the primary analysis, 19 [2.8%] patients experienced 1 other thrombovascular event (TVE) each (12 [3.5%] in the epoetin alfa group and 7 [2.1%] in the SOC group).
- Increased mortality was observed in a randomized, placebo-controlled study of epoetin alfa in adult patients who were undergoing CABG surgery (7 deaths in 126 patients randomized to epoetin alfa versus no deaths among 56 patients receiving placebo). Four of these deaths occurred during the period of study drug administration and all 4 deaths were associated with thrombotic events.
Prescribing and Distribution Program for darbepoetin alfa in Patients With Cancer
- In order to prescribe and/or dispense darbepoetin alfa to patients with cancer and anemia due to myelosuppressive chemotherapy, prescribers and hospitals must enroll in and comply with the ESA APPRISE Oncology Program requirements. To enroll, visit www.esa-apprise.com or call 1-866-284-8089 for further assistance. Additionally, prior to each new course of darbepoetin alfa in patients with cancer, prescribers and patients must provide written acknowledgment of a discussion of the risks of darbepoetin alfa.
Increased Mortality and/or Increased Risk of Tumor Progression or Recurrence in Patients With Cancer
- ESAs resulted in decreased locoregional control/progression-free survival and/or overall survival (see Table 3). These findings were observed in studies of patients with advanced head and neck cancer receiving radiation therapy (Studies 5 and 6), in patients receiving chemotherapy for metastatic breast cancer (Study 1) or lymphoid malignancy (Study 2), and in patients with non-small cell lung cancer or various malignancies who were not receiving chemotherapy or radiotherapy (Studies 7 and 8).

This image is provided by the National Library of Medicine.

Decreased Overall Survival
- Study 1 was described in the previous section. Mortality at 4 months (8.7% vs. 3.4%) was significantly higher in the epoetin alfa arm. The most common investigator-attributed cause of death within the first 4 months was disease progression; 28 of 41 deaths in the epoetin alfa arm and 13 of 16 deaths in the placebo arm were attributed to disease progression. Investigator-assessed time to tumor progression was not different between the 2 groups. Survival at 12 months was significantly lower in the epoetin alfa arm (70% vs. 76%; HR 1.37, 95% CI: 1.07, 1.75; p = 0.012).
- Study 2 was a randomized, double-blind study (darbepoetin alfa vs. placebo) conducted in 344 anemic patients with lymphoid malignancy receiving chemotherapy. With a median follow-up of 29 months, overall mortality rates were significantly higher among patients randomized to darbepoetin alfa as compared to placebo (HR 1.36, 95% CI: 1.02, 1.82).
- Study 7 was a multicenter, randomized, double-blind study (epoetin alfa vs. placebo) in which patients with advanced non-small cell lung cancer receiving only palliative radiotherapy or no active therapy were treated with epoetin alfa to achieve and maintain hemoglobin levels between 12 and 14 g/dL. Following an interim analysis of 70 patients (planned accrual 300 patients), a significant difference in survival in favor of the patients in the placebo arm of the study was observed (median survival 63 vs. 129 days; HR 1.84; p = 0.04).
- Study 8 was a randomized, double-blind study (darbepoetin alfa vs. placebo) in 989 anemic patients with active malignant disease, neither receiving nor planning to receive chemotherapy or radiation therapy. There was no evidence of a statistically significant reduction in proportion of patients receiving RBC transfusions. The median survival was shorter in the darbepoetin alfa treatment group than in the placebo group (8 months vs. 10.8 months; HR 1.30, 95% CI: 1.07, 1.57).
Decreased Progression-free Survival and Overall Survival
- Study 3 was a randomized, open-label, controlled, factorial design study in which darbepoetin alfa was administered to prevent anemia in 733 women receiving neo-adjuvant breast cancer treatment. A final analysis was performed after a median follow-up of approximately 3 years. The 3-year survival rate was lower (86% vs. 90%; HR 1.42, 95% CI: 0.93, 2.18) and the 3-year relapse-free survival rate was lower (72% vs. 78%; HR 1.33, 95% CI: 0.99, 1.79) in the darbepoetin alfa-treated arm compared to the control arm.
- Study 4 was a randomized, open-label, controlled study that enrolled 114 of a planned 460 cervical cancer patients receiving chemotherapy and radiotherapy. Patients were randomized to receive epoetin alfa to maintain hemoglobin between 12 and 14 g/dL or to RBC transfusion support as needed. The study was terminated prematurely due to an increase in thromboembolic adverse reactions in epoetin alfa-treated patients compared to control (19% vs. 9%). Both local recurrence (21% vs. 20%) and distant recurrence (12% vs. 7%) were more frequent in epoetin alfa-treated patients compared to control. Progression-free survival at 3 years was lower in the epoetin alfa-treated group compared to control (59% vs. 62%; HR 1.06, 95% CI: 0.58, 1.91). Overall survival at 3 years was lower in the epoetin alfa-treated group compared to control (61% vs. 71%; HR 1.28, 95% CI: 0.68, 2.42).
- Study 5 was a randomized, placebo-controlled study in 351 head and neck cancer patients where epoetin beta or placebo was administered to achieve target hemoglobins ≥ 14 and ≥ 15 g/dL for women and men, respectively. Locoregional progression-free survival was significantly shorter in patients receiving epoetin beta (HR 1.62, 95% CI: 1.22, 2.14; p = 0.0008) with medians of 406 days and 745 days in the epoetin beta and placebo arms respectively. Overall survival was significantly shorter in patients receiving epoetin beta (HR 1.39, 95% CI: 1.05, 1.84; p = 0.02).
Decreased Locoregional Control
- Study 6 was a randomized, open-label, controlled study conducted in 522 patients with primary squamous cell carcinoma of the head and neck receiving radiation therapy alone (no chemotherapy) who were randomized to receive darbepoetin alfa to maintain hemoglobin levels of 14 to15.5 g/dL or no darbepoetin alfa. An interim analysis performed on 484 patients demonstrated that locoregional control at 5 years was significantly shorter in patients receiving darbepoetin alfa (RR 1.44, 95% CI: 1.06, 1.96; p = 0.02). Overall survival was shorter in patients receiving darbepoetin alfa (RR 1.28, 95% CI: 0.98, 1.68; p = 0.08).
Hypertension
- Darbepoetin alfa is contraindicated in patients with uncontrolled hypertension. In darbepoetin alfa clinical studies, approximately 40% of patients with CKD required initiation or intensification of antihypertensive therapy during the early phase of treatment. Hypertensive encephalopathy and seizures have been reported in patients with CKD receiving darbepoetin alfa.
- Appropriately control hypertension prior to initiation of and during treatment with darbepoetin alfa. Reduce or withhold darbepoetin alfa if blood pressure becomes difficult to control. Advise patients of the importance of compliance with antihypertensive therapy and dietary restrictions.
Seizures
- Darbepoetin alfa increases the risk of seizures in patients with CKD. During the first several months following initiation of darbepoetin alfa, monitor patients closely for premonitory neurologic symptoms. Advise patients to contact their healthcare practitioner for new-onset seizures, premonitory symptoms, or change in seizure frequency.
Lack or Loss of Hemoglobin Response to darbepoetin alfa
- For lack or loss of hemoglobin response to darbepoetin alfa, initiate a search for causative factors (e.g., iron deficiency, infection, inflammation, bleeding). If typical causes of lack or loss of hemoglobin response are excluded, evaluate for PRCA.
- In the absence of PRCA, follow dosing recommendations for management of patients with an insufficient hemoglobin response to darbepoetin alfa therapy.
Pure Red Cell Aplasia
- Cases of PRCA and of severe anemia, with or without other cytopenias that arise following the development of neutralizing antibodies to erythropoietin have been reported in patients treated with darbepoetin alfa. This has been reported predominantly in patients with CKD receiving ESAs by subcutaneous administration. PRCA has also been reported in patients receiving ESAs for anemia related to hepatitis C treatment (an indication for which darbepoetin alfa is not approved).
- If severe anemia and low reticulocyte count develop during treatment with darbepoetin alfa, withhold darbepoetin alfa and evaluate patients for neutralizing antibodies to erythropoietin. Contact Amgen (1-800-77-AMGEN) to perform assays for binding and neutralizing antibodies. Permanently discontinue darbepoetin alfa in patients who develop PRCA following treatment with darbepoetin alfa or other erythropoietin protein drugs. Do not switch patients to other ESAs.
Serious Allergic Reactions
- Serious allergic reactions, including anaphylactic reactions, angioedema, bronchospasm, skin rash, and urticaria may occur with darbepoetin alfa. Immediately and permanently discontinue darbepoetin alfa and administer appropriate therapy if a serious allergic or anaphylactic reaction occurs.
Dialysis Management
- Patients may require adjustments in their dialysis prescriptions after initiation of darbepoetin alfa. Patients receiving darbepoetin alfa may require increased anticoagulation with heparin to prevent clotting of the extracorporeal circuit during hemodialysis.
Laboratory Monitoring
- Evaluate transferrin saturation and serum ferritin prior to and during darbepoetin alfa treatment. Administer supplemental iron therapy when serum ferritin is less than 100 mcg/L or when serum transferrin saturation is less than 20%. * The majority of patients with CKD will require supplemental iron during the course of ESA therapy. Following initiation of therapy and after each dose adjustment, monitor hemoglobin weekly until the hemoglobin is stable and sufficient to minimize the need for RBC transfusion. Thereafter, hemoglobin may be monitored less frequently provided hemoglobin levels remain stable.
Adverse Reactions
Clinical Trials Experience
- The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism.
- Increased mortality and/or increased risk of tumor progression or recurrence in Patients With Cancer.
- Hypertension.
- Seizures.
- PRCA
- Serious allergic reactions.
Clinical Trial Experience
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of other drugs and may not reflect the rates observed in practice.
Patients with Chronic Kidney Disease
Adult Patients
- Adverse reactions were determined based on pooled data from 5 randomized, active-controlled studies of darbepoetin alfa with a total of 1357 patients (darbepoetin alfa 766, epoetin alfa 591). The median duration of exposure for patients receiving darbepoetin alfa was 340 days, with 580 patients exposed for greater than 6 months and 360 patients exposed for greater than 1 year. The median (25th, 75th percentiles) weight-adjusted dose of darbepoetin alfa was 0.50 mcg/kg (0.32, 0.81). The median (range) age for patients administered darbepoetin alfa was 62 years (18 to 88). In the darbepoetin alfa group, 55% were male, 72% were white, 83% were receiving dialysis, and 17% were not receiving dialysis.
- Table 4 lists adverse reactions occurring in ≥ 5% of patients treated with darbepoetin alfa.

This image is provided by the National Library of Medicine.

- Rates of adverse reactions with darbepoetin alfa therapy were similar to those observed with other recombinant erythropoietins in these studies.
Pediatric Patients
- Darbepoetin alfa was administered to 81 pediatric patients with CKD who had stable hemoglobin concentrations while previously receiving epoetin alfa. In this study, the most frequently reported serious adverse reactions with darbepoetin alfa were hypertension and convulsions. The most commonly reported adverse reactions were hypertension, injection site pain, rash, and convulsions. Darbepoetin alfa administration was discontinued because of injection site pain in 2 patients and moderate hypertension in a third patient.
- Studies have not evaluated the effects of darbepoetin alfa when administered to pediatric patients as the initial treatment for the anemia associated with CKD.
Cancer Patients Receiving Chemotherapy
- Adverse reactions were based on data from a randomized, double-blind, placebo-controlled study of darbepoetin alfa in 597 patients (darbepoetin alfa 301, placebo 296) with extensive stage small cell lung cancer (SCLC) receiving platinum-based chemotherapy. All patients were white, 64% were male, and the median age was 61 years (range: 28 to 82 years); 25% of the study population were from North America, Western Europe, and Australia. Patients received darbepoetin alfa at a dose of 300 mcg or placebo weekly for 4 weeks then every 3 weeks for a total of 24 weeks, and the median duration of exposure was 19 weeks (range: 1 to 26 weeks).
- Adverse reactions were also based on data from 7 randomized, double-blind, placebo-controlled studies, including the SCLC study described above, that enrolled 2112 patients (darbepoetin alfa 1203, placebo 909) with non-myeloid malignancies. Most patients were white (95%), male (52%), and the median age was 63 years (range: 18 to 91 years); 73% of the study population were from North America, Western Europe, and Australia. Dosing and schedules varied by study from once weekly to once every 4 weeks, and the median duration of exposure was 12 weeks (range: 1 to 27 weeks).

This image is provided by the National Library of Medicine.

- In addition to the thrombovascular adverse reactions, abdominal pain and edema occurred at a higher incidence in patients taking darbepoetin alfa compared to patients on placebo. Among all placebo-controlled studies, abdominal pain (13.2% vs. 9.4%) and edema (12.8% vs. 9.7%) were reported more frequently in patients receiving darbepoetin alfa compared to the placebo group. In the SCLC study the incidence of abdominal pain (10.3% vs. 3.4%) and edema (5.6% vs. 5.1%) in the darbepoetin alfa-treated patients compared to those receiving placebo.
Postmarketing Experience
Postmarketing Experience
- Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- The following adverse reactions have been identified during postmarketing use of darbepoetin alfa:
- Seizures
- PRCA
- Serious allergic reactions
Immunogenicity
- As with all therapeutic proteins, there is a potential for immunogenicity. Neutralizing antibodies to darbepoetin alfa that cross-react with endogenous erythropoietin and other ESAs can result in PRCA or severe anemia (with or without other cytopenias).
- In clinical studies, the percentage of patients with antibodies to darbepoetin alfa was examined using the Biacore® assay. Sera from 1501 patients with CKD and 1159 cancer patients were tested. At baseline, prior to darbepoetin alfa treatment, binding antibodies were detected in 59 patients (4%) with CKD and 36 cancer patients (3%). During darbepoetin alfa therapy (range: 22 to 177 weeks), a follow-up sample was taken. One additional patient with CKD and 8 additional cancer patients developed antibodies capable of binding darbepoetin alfa. None of the patients had antibodies capable of neutralizing the activity of darbepoetin alfa or endogenous erythropoietin at baseline or at end of study. No clinical sequelae consistent with PRCA were associated with the presence of these antibodies.
- The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to darbepoetin alfa with the incidence of antibodies to other products may be misleading.
Drug Interactions
There is limited information regarding Darbepoetin alfa Drug Interactions in the drug label.
Use in Specific Populations
Pregnancy
- There are no adequate and well-controlled studies of darbepoetin alfa use in pregnant women. In animal reproduction and developmental toxicity studies, darbepoetin alfa increased early post-implantation loss. Use darbepoetin alfa during pregnancy only if the potential benefit justifies the potential risk to the fetus.
- When darbepoetin alfa was administered intravenously to healthy pregnant rats and rabbits, there was no evidence of embryofetal toxicity or other adverse outcomes at the intravenous doses tested, up to 20 mcg/kg/day. This animal dose level of 20 mcg/kg/day is approximately 20-fold higher than the clinical recommended starting dose, depending on the patient’s treatment indication. Slightly reduced fetal weights were observed when healthy rat and rabbit mothers received doses of 1 mcg/kg or more. This dose of 1 mcg/kg is near the clinical recommended starting dose. While no adverse effects on uterine implantation occurred in animals, there was an increase in early post-implantation loss in animal fertility studies. It is not clear whether the increased post-implantation loss reflects a drug effect on the uterine environment or on the conceptus. No significant placental transfer of darbepoetin alfa was detected.
- In a peri/postnatal development study, pregnant female rats received darbepoetin alfa intravenously every other day from implantation throughout pregnancy and lactation. The lowest dose tested, 0.5 mcg/kg, did not cause fetal toxicity; this dose is approximately equivalent to the clinical recommended starting dose. At maternal doses of 2.5 mcg/kg and higher, pups had decreased fetal body weights, which correlated with a slight increase in the incidence of fetal deaths, as well as delayed eye opening and delayed preputial separation.
- Women who become pregnant during darbepoetin alfa treatment are encouraged to enroll in Amgen’s Pregnancy Surveillance P.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Darbepoetin alfa in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Darbepoetin alfa during labor and delivery.
Nursing Mothers
- It is not known whether darbepoetin alfa is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when darbepoetin alfa is administered to a nursing woman.
Pediatric Use
- Darbepoetin alfa safety and efficacy were similar between adults and pediatric patients with CKD who were over 1 year of age when patients were transitioned from treatment with epoetin alfa to darbepoetin alfa.
- Darbepoetin alfa safety and efficacy have not been established in the initial treatment of anemic pediatric patients with CKD or in the transition from another erythropoietin to darbepoetin alfa in pediatric CKD patients less than 1 year of age.
Pediatric Cancer Patients
- The safety and efficacy of darbepoetin alfa in pediatric cancer patients have not been established.
Geriatic Use
- Of the 1801 patients with CKD in clinical studies of darbepoetin alfa, 44% were age 65 and over, while 17% were age 75 and over. Of the 873 patients in clinical studies receiving darbepoetin alfa and concomitant cancer chemotherapy, 45% were age 65 and over, while 14% were age 75 and over. No differences in safety or efficacy were observed between older and younger patients.
Gender
There is no FDA guidance on the use of Darbepoetin alfa with respect to specific gender populations.
Race
There is no FDA guidance on the use of Darbepoetin alfa with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Darbepoetin alfa in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Darbepoetin alfa in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Darbepoetin alfa in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Darbepoetin alfa in patients who are immunocompromised.
Administration and Monitoring
Administration
Monitoring
- To undergo regular blood pressure monitoring, adhere to prescribed anti-hypertensive regimen and follow recommended dietary restrictions.
- Evaluate transferrin saturation and serum ferritin prior to and during darbepoetin alfa treatment. Administer supplemental iron therapy when serum ferritin is less than 100 mcg/L or when serum transferrin saturation is less than 20%. The majority of patients with CKD will require supplemental iron during the course of ESA therapy.
- Following initiation of therapy and after each dose adjustment, monitor hemoglobin weekly until the hemoglobin is stable and sufficient to minimize the need for RBC transfusion. Thereafter, hemoglobin may be monitored less frequently provided hemoglobin levels remain stable.
IV Compatibility
There is limited information regarding IV Compatibility of Darbepoetin alfa in the drug label.
Overdosage
- Darbepoetin alfa overdosage can cause hemoglobin levels above the desired level, which should be managed with discontinuation or reduction of darbepoetin alfa dosage and/or with phlebotomy, as clinically indicated. Cases of severe hypertension have been observed following overdose with ESAs
Pharmacology
There is limited information regarding Darbepoetin alfa Pharmacology in the drug label.
Mechanism of Action
- Darbepoetin alfa stimulates erythropoiesis by the same mechanism as endogenous erythropoetin.
Structure
- Darbepoetin alfa is an erythropoiesis-stimulating protein that is produced in Chinese hamster ovary (CHO) cells by recombinant DNA technology.
- Darbepoetin alfa is a 165-amino acid protein that differs from recombinant human erythropoietin in containing 5 N-linked oligosaccharide chains, whereas recombinant human erythropoietin contains 3 chains. The 2 additional N-glycosylation sites result from amino acid substitutions in the erythropoietin peptide backbone. The approximate molecular weight of darbepoetin alfa is 37,000 daltons.
- Darbepoetin alfa is formulated as a sterile, colorless, preservative-free solution containing polysorbate for intravenous or subcutaneous administration. Each 1 mL contains polysorbate 80 (0.05 mg), sodium chloride (8.18 mg), sodium phosphate dibasic anhydrous (0.66 mg), and sodium phosphate monobasic monohydrate (2.12 mg) in Water for Injection, USP (pH 6.2 ± 0.2).
Pharmacodynamics
- Increased hemoglobin levels are not generally observed until 2 to 6 weeks after initiating treatment with darbepoetin alfa.
Pharmacokinetics
Adult Patients with CKD
- The pharmacokinetics of darbepoetin alfa were studied in patients with CKD receiving or not receiving dialysis and cancer patients receiving chemotherapy.
- Following intravenous administration of darbepoetin alfa to patients with CKD receiving dialysis, darbepoetin alfa serum concentration-time profiles were biphasic, with a distribution half-life of approximately 1.4 hours and a mean terminal half-life (t1/2) of 21 hours. The t1/2 of darbepoetin alfa was approximately 3-fold longer than that of epoetin alfa when administered intravenously.
- Following subcutaneous administration of darbepoetin alfa to patients with CKD (receiving or not receiving dialysis), absorption was slow and Cmax occurred at 48 hours (range: 12 to 72 hours). In patients with CKD receiving dialysis, the average t1/2 was 46 hours (range: 12 to 89 hours), and in patients with CKD not receiving dialysis, the average t1/2 was 70 hours (range: 35 to 139 hours). Darbepoetin alfa apparent clearance was approximately 1.4 times faster on average in patients receiving dialysis compared to patients not receiving dialysis. The bioavailability of darbepoetin alfa in patients with CKD receiving dialysis after subcutaneous administration was 37% (range: 30% to 50%).
Pediatric Patients with CKD
- Darbepoetin alfa pharmacokinetics was studied in 12 pediatric patients (age 3 to 16 years) with CKD receiving or not receiving dialysis. Following a single intravenous or subcutaneous darbepoetin alfa dose, Cmax and t1/2 were similar to those obtained in adult patients with CKD on dialysis. Following a single subcutaneous dose, the average bioavailability was 54% (range: 32% to 70%), which was higher than that obtained in adult patients with CKD on dialysis.
Adult Cancer Patients
- Following the first subcutaneous dose of 6.75 mcg/kg (equivalent to 500 mcg for a 74-kg patient) in patients with cancer, the mean t1/2 was 74 hours (range: 24 to 144 hours) and Cmax was observed at 71 hours (range: 28 to 120 hours). When administered on a once every 3 week schedule, 48-hour postdose darbepoetin alfa levels after the fourth dose were similar to those after the first dose.
- Over the dose range of 0.45 to 4.5 mcg/kg darbepoetin alfa administered intravenously or subcutaneously on a once weekly schedule and 4.5 to 15 mcg/kg administered subcutaneously on a once every 3 week schedule, systemic exposure was approximately proportional to dose. No evidence of accumulation was observed beyond an expected less than 2-fold increase in blood levels when compared to the initial dose.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
- The carcinogenic potential of darbepoetin alfa has not been evaluated in long-term animal studies. In toxicity studies of approximately 6 months duration in rats and dogs, no tumorigenic or unexpected mitogenic responses were observed in any tissue type.
Mutagenicity
- Darbepoetin alfa was not mutagenic or clastogenic under the conditions tested. Darbepoetin alfa was negative in the in vitro bacterial reverse mutation assay, the in vitro mammalian cell gene mutation assay (using CHO cells), and in the in vivo mouse erythrocyte micronucleus assay.
Impairment of Fertility
- Darbepoetin alfa increased the incidence of post-implantation losses in rats. Male and female rats received intravenous doses prior to and during mating; then females were treated 3 times weekly during the first trimester of gestation (gestation days 1, 3, 5, and 7). No effect on reproductive performance, fertility, or sperm assessment parameters were detected at any of the doses evaluated (up to 10 mcg/kg, administered 3 times weekly). The dose of 10 mcg/kg is more than 10-fold higher than the clinical recommended starting dose. An increase in post-implantation fetal loss was seen at doses equal to or greater than 0.5 mcg/kg, administered 3 times weekly. The dose of 0.5 mcg/kg is approximately equivalent to the clinical recommended starting dose. Signs of exaggerated pharmacology were not observed in the mother receiving 0.5 mcg/kg or less, but were observed at 2.5 mcg/kg and higher.
Reproductive and Developmental Toxicology
- When darbepoetin alfa was administered intravenously during organogenesis to pregnant rats (gestational days 6 to 15) and rabbits (gestational days 6 to 18), no evidence of direct embryotoxic, fetotoxic, or teratogenic outcomes were observed at the doses tested, up to 20 mcg/kg/day. This animal dose level of 20 mcg/kg/day is approximately 20-fold higher than the clinical recommended starting dose, depending on the patient’s treatment indication. The only adverse effect observed was a slight reduction in fetal weight, which occurred only at doses causing exaggerated pharmacological effects in both the rat and rabbit dams (1 mcg/kg/day and higher). No deleterious effects on uterine implantation were seen in either species.
- No significant placental transfer of darbepoetin alfa was observed in rats; placental transfer was not evaluated in rabbits.
- In a peri/postnatal development study, pregnant female rats were treated intravenously with darbepoetin alfa day 6 of gestation through day 23 of lactation at 2.5 mcg/kg and higher every other day. Pups of treated mothers had decreased fetal body weights, which correlated with slight increases in the incidences of fetal death, as well as delayed eye opening and delayed preputial separation. The offspring (F1 generation) of the treated rats were observed postnatally; rats from the F1 generation reached maturity and were mated; no darbepoetin alfa-related effects were apparent for their offspring (F2 generation fetuses).
Clinical Studies
- Clinical studies in the nephrology and chemotherapy-induced anemia clinical programs are designated with the prefixes “N” and “C”, respectively.
Patients With Chronic Kidney Disease
- Patients with chronic kidney disease on dialysis: ESA effects on rates of transfusion
- In early clinical studies conducted in CKD patients on dialysis, ESAs have been shown to reduce the use of RBC transfusions. These studies enrolled patients with mean baseline hemoglobin levels of approximately 7.5 g/dL and ESAs were generally titrated to achieve a hemoglobin level of approximately 12 g/dL. Fewer transfusions were given during the ESA treatment period when compared to a pre-treatment interval.
- In the Normal Hematocrit Study, the yearly transfusion rate was 51.5% in the lower hemoglobin group (10 g/dL) and 32.4% in the higher hemoglobin group (14 g/dL).
- Patients with chronic kidney disease not on dialysis: ESA effects on rates of transfusion.
- In TREAT, a randomized, double-blind trial of 4038 patients with CKD and type 2 diabetes not on dialysis, a post-hoc analysis showed that the proportion of patients receiving RBC transfusions was lower in patients administered darbepoetin alfa to target a hemoglobin of 13 g/dL compared to the control arm in which darbepoetin alfa was administered intermittently if hemoglobin concentration decreased to less than 9 g/dL (15% versus 25%, respectively). In CHOIR, a randomized open-label study of 1432 patients with CKD not on dialysis, use of an ESA to target a higher (13.5 g/dL) versus lower (11.3 g/dL) hemoglobin goal did not reduce the use of RBC transfusions. In each trial, no benefits occurred for the cardiovascular or end-stage renal disease outcomes. In each trial, the potential benefit of ESA therapy was offset by worse cardiovascular safety outcomes resulting in an unfavorable benefit-risk profile.
ESA Effects on quality of life
- Darbepoetin alfa use has not been demonstrated in controlled clinical trials to improve quality of life, fatigue, or patient well-being.
- ESA Effects on rates of death and other serious cardiac adverse events
- Three randomized outcome trials (Normal Hematocrit Study [NHS], Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease [CHOIR], and Trial of Darbepoetin Alfa in Type 2 Diabetes and CKD [TREAT]) have been conducted in patients with CKD using Epogen/PROCRIT/darbepoetin alfa to target higher vs. lower hemoglobin levels. Though these trials were designed to establish a cardiovascular or renal benefit of targeting higher hemoglobin levels, in all 3 studies, patients randomized to the higher hemoglobin target experienced worse cardiovascular outcomes and showed no reduction in progression to ESRD. In each trial, the potential benefit of ESA therapy was offset by worse cardiovascular safety outcomes resulting in an unfavorable benefit-risk profile.
Other ESA trials
- Two studies evaluated the safety and efficacy of the de novo use of darbepoetin alfa for the correction of anemia in adult patients with CKD, and 3 studies (2 in adults and 1 in pediatric patients) assessed the ability of darbepoetin alfa to maintain hemoglobin concentrations in patients with CKD who had been receiving other recombinant erythropoietins.
De Novo Use of darbepoetin alfa
- Once Weekly darbepoetin alfa Starting Dose
- In 2 randomized, open-label studies, darbepoetin alfa or epoetin alfa was administered for the correction of anemia in patients with CKD who had not been receiving prior treatment with exogenous erythropoietin. Study N1 evaluated CKD patients receiving dialysis; Study N2 evaluated patients not requiring dialysis. In both studies, the starting dose of darbepoetin alfa was 0.45 mcg/kg administered once weekly. The starting dose of epoetin alfa was 50 Units/kg 3 times weekly in Study N1 and 50 Units/kg twice weekly in Study N2. When necessary, dosage adjustments were instituted to maintain hemoglobin in the study target range of 11 to 13 g/dL. (Note: The recommended hemoglobin target range is lower than the target range of these studies. The primary efficacy endpoint was the proportion of patients who experienced at least a 1 g/dL increase in hemoglobin concentration to a level of at least 11 g/dL by 20 weeks (Study N1) or 24 weeks (Study N2). The studies were designed to assess the safety and effectiveness of darbepoetin alfa but not to support conclusions regarding comparisons between the 2 products.
- In Study N1, the primary efficacy endpoint was achieved by 72% (95% CI: 62%, 81%) of the 90 patients treated with darbepoetin alfa and 84% (95% CI: 66%, 95%) of the 31 patients treated with epoetin alfa. The mean increase in hemoglobin over the initial 4 weeks of darbepoetin alfa treatment was 1.1 g/dL (95% CI: 0.82 g/dL, 1.37 g/dL).
- In Study N2, the primary efficacy endpoint was achieved by 93% (95% CI: 87%, 97%) of the 129 patients treated with darbepoetin alfa and 92% (95% CI: 78%, 98%) of the 37 patients treated with epoetin alfa. The mean increase in hemoglobin from baseline through the initial 4 weeks of darbepoetin alfa treatment was 1.38 g/dL (95% CI: 1.21 g/dL, 1.55 g/dL).
Once Every 2 Week darbepoetin alfa Starting Dose
- In 2 single-arm studies (N3 and N4), darbepoetin alfa was administered for the correction of anemia in CKD patients not receiving dialysis. In both studies, the starting dose of darbepoetin alfa was 0.75 mcg/kg administered once every 2 weeks.
- In Study N3 (study duration of 18 weeks), the hemoglobin goal (hemoglobin concentration ≥ 11 g/dL) was achieved by 92% (95% CI: 86%, 96%) of the 128 patients treated with darbepoetin alfa.
- In Study N4 (study duration of 24 weeks), the hemoglobin goal (hemoglobin concentration of 11 to 13 g/dL) was achieved by 85% (95% CI: 77%, 93%) of the 75 patients treated with darbepoetin alfa.
Conversion from Other Recombinant Erythropoietins
- Two studies of adults (N5 and N6) and 1 study in pediatric patients (N7) were conducted in patients who had been receiving other recombinant erythropoietins for treatment of the anemia due to CKD. The studies compared the abilities of darbepoetin alfa and other erythropoietins to maintain hemoglobin concentrations within a study target range of 9 to 13 g/dL in adults and 10 to 12.5 g/dL in pediatric patients. (Note: The recommended hemoglobin target is lower than the target range of these studies) . Patients who had been receiving stable doses of other recombinant erythropoietins were randomized to darbepoetin alfa or continued with their prior erythropoietin at the previous dose and schedule. For patients randomized to darbepoetin alfa, the initial weekly dose was determined on the basis of the previous total weekly dose of recombinant erythropoietin.
Adult Patients
- Study N5 was a double-blind study in which 169 hemodialysis patients were randomized to treatment with darbepoetin alfa and 338 patients continued on epoetin alfa. * Study N6 was an open-label study in which 347 patients were randomized to treatment with darbepoetin alfa and 175 patients were randomized to continue on epoetin alfa or epoetin beta. Of the patients randomized to darbepoetin alfa, 92% were receiving hemodialysis and 8% were receiving peritoneal dialysis.
- In Study N5, a median weekly dose of 0.53 mcg/kg darbepoetin alfa (25th, 75th percentiles: 0.30, 0.93 mcg/kg) was required to maintain hemoglobin in the study target range. In Study N6, a median weekly dose of 0.41 mcg/kg darbepoetin alfa (25th, 75th percentiles: 0.26, 0.65 mcg/kg) was required to maintain hemoglobin in the study target range.
Pediatric Patients
- Study N7 was an open-label, randomized study conducted in the United States in pediatric patients from 1 to 18 years of age with CKD receiving or not receiving dialysis. Eighty-one patients with hemoglobin concentrations that were stable on epoetin alfa received darbepoetin alfa (subcutaneously or intravenously), and 42 patients continued to receive epoetin alfa at the current dose, schedule, and route of administration. Patients received darbepoetin alfa once weekly if previously receiving epoetin alfa 2 or 3 times weekly or once every other week if previously receiving epoetin alfa weekly. A median weekly dose of 0.41 mcg/kg darbepoetin alfa (25th, 75th percentiles: 0.25, 0.82 mcg/kg) was required to maintain hemoglobin in the study target range.
Cancer Patients Receiving Chemotherapy
- The safety and efficacy of darbepoetin alfa was assessed in two multicenter, randomized studies in patients with anemia due to the effect of concomitantly administered cancer chemotherapy. Study C1 was a randomized (1:1), placebo-controlled, double-blind, multinational study conducted in 314 patients where darbepoetin alfa was administered weekly. Study C2 was a randomized (1:1), double-blind, double-dummy, active-controlled, multinational study conducted in 705 patients where darbepoetin alfa was administered either every week or every 3 weeks. Efficacy was demonstrated by a statistically significant reduction in the proportion of patients receiving RBC transfusions among patients who were on study therapy for more than 28 days.
Study C1
- Study C1 was conducted in anemic patients (hemoglobin ≤ 11 g/dL) with non-small cell lung cancer or small cell lung cancer who were scheduled to receive at least 12 weeks of a platinum-containing chemotherapy regimen. Randomization was stratified by tumor type and region (Australia vs. Canada vs. Europe). Patients received darbepoetin alfa 2.25 mcg/kg or placebo as a weekly subcutaneous injection commencing on the first day of the chemotherapy cycle. Efficacy was determined by a reduction in the proportion of patients who received RBC transfusions between week 5 (day 29) and end of treatment period (12 weeks) in the subset of 297 randomized patients (148 darbepoetin alfa and 149 placebo) who were on-study at the beginning of study week 5. All 297 patients were white, 72% were male, 71% had non-small cell histology, and the median age was 62 years (range: 36 to 80). A significantly lower proportion of patients in the darbepoetin alfa arm received RBC transfusions during week 5 to the end of treatment compared to patients in the placebo arm (crude percentages: 26% vs. 50%; p < 0.001, based on a comparison of the difference in Kaplan-Meier proportions using the Cochran-Mantel-Haenszel strata-adjusted Chi-square test).
Study C2
- Study C2 was conducted in anemic patients (hemoglobin < 11 g/dL) with non-myeloid malignancies receiving chemotherapy. Randomization was stratified by region (Western vs. Central/Eastern Europe), tumor type (lung and gynecological vs. others), and baseline hemoglobin (< 10 vs. ≥ 10 g/dL); all patients received double-dummy placebo and either darbepoetin alfa 500 mcg every 3 weeks or darbepoetin alfa 2.25 mcg/kg weekly subcutaneous injections for 15 weeks. Only 1 patient was non-white, 55% were female, and the median age was 60 years (range: 20 to 86). One hundred seven patients (16%) had lung or gynecological cancer while 565 (84%) had other tumor types. In both treatment schedules, the dose was reduced by 40% of the previous dose if hemoglobin level increased by more than 1 g/dL in a 14-day period.
- Efficacy was determined by a comparison of the proportion of patients who received at least 1 RBC transfusion between week 5 (day 29) and the end of treatment. Three hundred thirty-five patients in the every 3 week dosing arm and 337 patients in the weekly dosing arm remained on study through or beyond day 29 and were evaluable for efficacy. Two hundred thirty-eight patients (71%) in the every 3 week arm and 261 patients (77%) patients in the weekly arm required dose reductions. Twenty-three percent (95% CI: 18%, 28%) of patients in the every 3 week treatment schedule and 28% (95% CI: 24%, 34%) in the weekly schedule received at least 1 RBC transfusion. The observed difference in the RBC transfusion rates (every 3 week minus weekly) was -5.8% (95% CI: -12.4%, 0.8%).
Study C3
Lack of Efficacy in Improving Survival
- Study C3 was conducted in patients required to have a hemoglobin concentration ≥ 9 g/dL and ≤ 13 g/dL with previously untreated extensive-stage small cell lung cancer (SCLC) receiving platinum and etoposide chemotherapy. Randomization was stratified by region (Western Europe, Australia/North America, and rest of world), Eastern Cooperative Oncology Group (ECOG) performance status (0 or 1 vs. 2), and lactate dehydrogenase (below vs. above the upper limit of normal). Patients were randomized to receive darbepoetin alfa (n = 298) at a dose of 300 mcg once weekly for the first 4 weeks, followed by 300 mcg once every 3 weeks for the remainder of the treatment period or placebo (n = 298).
- This study was designed to detect a prolongation in overall survival (from a median of 9 months to a median of 12 months). For the final analysis, there was no evidence of improved survival (p=0.43, log-rank test).
How Supplied
There is limited information regarding Darbepoetin alfa How Supplied in the drug label.
Storage
- Store at 36°F to 46°F (2°C to 8°C). Do not freeze.
Images
Drug Images
Package and Label Display Panel













Patient Counseling Information
- Prior to treatment, inform patients of the risks and benefits of Aranesp.
- Inform patients with cancer that they must sign the patient-healthcare provider acknowledgment form before the start of each treatment course with Aranesp and that healthcare providers must enroll and comply with the ESA APPRISE Oncology Program in order to prescribe Aranesp.
- Inform patients:
- To read the Medication Guide.
- Of the increased risks of mortality, serious cardiovascular reactions, thromboembolic reactions, stroke, and tumor progression.
- To undergo regular blood pressure monitoring, adhere to prescribed anti-hypertensive regimen and follow recommended dietary restrictions.
- To contact their healthcare provider for new-onset neurologic symptoms or change in seizure frequency.
- Of the need to have regular laboratory tests for hemoglobin.
- Instruct patients who self-administer Aranesp of the:
- Importance of following the Instructions for Use.
- Dangers of reusing needles, syringes, or unused portions of single-dose vials.
- Proper disposal of used syringes, needles, and unused vials, and of the full container.
Precautions with Alcohol
- Alcohol-Darbepoetin alfa interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- ARANESP ®[1]
Look-Alike Drug Names
There is limited information regarding Darbepoetin alfa Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.