Wilson's disease pathophysiology: Difference between revisions
| Line 56: | Line 56: | ||
</div> | </div> | ||
==Microscopic pathology== | ==Microscopic pathology== | ||
* Histological examination of a liver biopsy may show the following:<ref name="pmid167753002">{{cite journal| author=Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW et al.| title=MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. | journal=AJNR Am J Neuroradiol | year= 2006 | volume= 27 | issue= 6 | pages= 1373-8 | pmid=16775300 | doi= | pmc= | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=16775300 }}</ref> | |||
** Mild steatosis which is considered an early histological feature | |||
** Glycogenated hepatic nuclei | |||
** Hepatocellular necrosis | |||
** Autoimmune hepatitis histologic features | |||
** Fibrosis and cirrhosis (macronodular or micronodular) in advanced cases | |||
** Fulminant liver falilure features which include: | |||
*** Hepatocellular degenration | |||
*** Parenchymal collapse | |||
==References== | ==References== | ||
Revision as of 19:54, 20 December 2017
| https://https://www.youtube.com/watch?v=Cr8R_bnKAtk%7C350}} |
|
Wilson's disease Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Wilson's disease pathophysiology On the Web |
|
American Roentgen Ray Society Images of Wilson's disease pathophysiology |
|
Risk calculators and risk factors for Wilson's disease pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmed Elsaiey, MBBCH [2]
Overview
Pathophysiology
Normal copper transportation and metabolism
- Copper is one of the essential elements that is required daily in diet in a range of 1.5 to 2 mg.[1]
- Copper is incorporated in number of important functions in the body. It functions as a component of some enzymes as cytochrome c oxidase, dopamine β-hydroxylase, superoxide dismutase and tyrosinase.[2]
- In order to function appropriately, a significant proportion of Copper must be absorbed and transported to the different site of actions. Stomach and duodenum are the main sites of absorption of copper then it binds albumin and transported to different body tissues. The excess copper is excreted as feces after being a part of the bile.
- Copper enters the body through the digestive tract. A transporter protein on the cells of the small bowel, copper membrane transporter 1 (CMT1), carries copper inside the cells, where some is bound to metallothionein and part is carried by ATOX1 to an organelle known as the trans-Golgi network. Here, in response to rising concentrations of copper, an enzyme called ATP7A releases copper into the portal vein to the liver. Liver cells also carry the CMT1 protein, and metallothionein and ATOX1 bind it inside the cell, but here it is ATP7B that links copper to ceruloplasmin and releases it into the bloodstream, as well as removing excess copper by secreting it into bile. Both functions of ATP7B are impaired in Wilson's disease. Copper accumulates in the liver tissue; ceruloplasmin is still secreted, but in a form that lacks copper (termed apoceruloplasmin) and rapidly degraded in the bloodstream.
- Transportation of the copper to different body tissues is regulated by ATP7B gene in the hepatocytes.
- ATP7B gene acts on transporting copper via two ways:[3][4]
- In the trans-golgi network: Formation of ceruloplasmin (which is secreted into the blood) by adding the copper to the apoceruloplasmin.
- In the cytoplasmic vesicles: Excretion of the excess copper into the bile by exocytosis against the hepatocytes memebrane.

Pathogenesis
Genetics
The Wilson's disease gene (ATP7B) has been mapped to chromosome 13 (13q14.3) and is expressed primarily in the liver, kidney, and placenta. The gene codes for a P-type (cation transport enzyme) ATPase that transports copper into bile and incorporates it into ceruloplasmin. Mutations can be detected in 90% of patients. Most (60%) are homozygous for ATP7B mutations (two abnormal copies), and 30% have only one abnormal copy. 10% have no detectable mutation.
Although 300 mutations of ATP7B have been described, in most populations the cases of Wilson's disease are due to a small number of mutations specific for that population. For instance, in Western populations the H1069Q mutation (replacement of a histidineby a glutamine at position 1069 in the gene) is present in 37-63% of cases, while in China this mutation is very uncommon and R778L (arginine to leucine at 778) is found more often. Relatively little is known about the relative impact of various mutations, although the H1069Q mutation seems to predict later onset and predominantly neurological problems, according to some studies.[5]
A normal variation in the PRNP gene can modify the course of the disease by delaying the age of onset and affecting the type of symptoms that develop. This gene produces prion protein, which is active in the brain and other tissues and also appears to be involved in transporting copper.[6] A role for the ApoE gene was initially suspected but could not be confirmed.[5]
The condition is inherited in an autosomal recessive pattern, which means both copies of the gene have mutations. In order to inherit it, both of the parents of an individual must carry an affected gene. Most patients have no family history of the condition.[5] People with only one abnormal gene are called carriers (heterozygotes) and may have mild, but medically insignificant, abnormalities of copper metabolism.
Wilson's disease is the most common of a group of hereditary diseases that cause copper overload in the liver. All can cause cirrhosis at a young age. The other members of the group are Indian childhood cirrhosis (ICC), endemic Tyrolean infantile cirrhosis and idiopathic copper toxicosis. These are not related to ATP7B mutations, but ICC has been linked to mutations in the KRT8 and the KRT18 gene.[5]
Other species
Hereditary copper accumulation has been described in Bedlington Terriers,[7] where it generally only affects the liver. It is due to mutations in the COMMD1 (or MURR1) gene.[8] In patients with non-Wilsonian copper accumulation states (such as Indian childhood cirrhosis), no COMMD1 mutations could be detected to explain their genetic origin.[9]
Associated conditions
Gross pathology
-
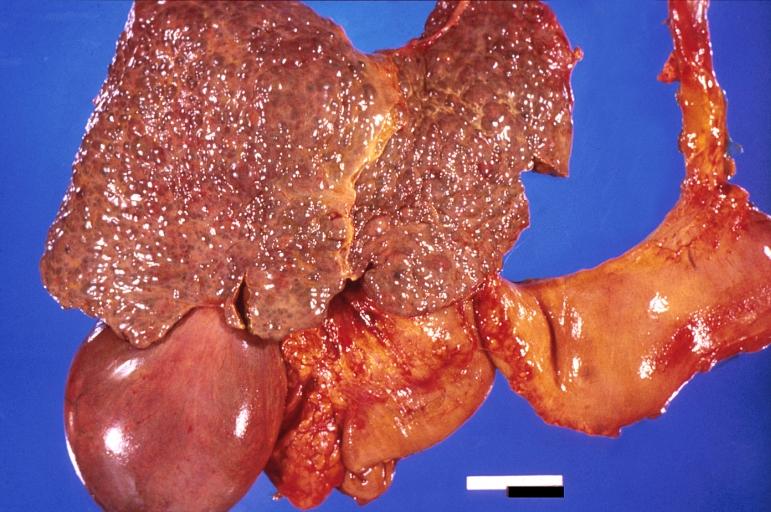
Liver cirrhosis and enlarged gall bladder in Wilson's disease.
-
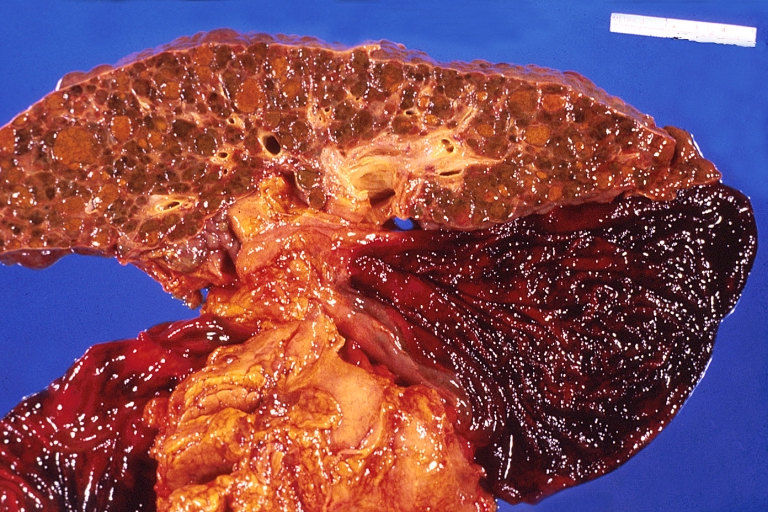
Liver cirrhosis and enlarged gall bladder in Wilson's disease.
-
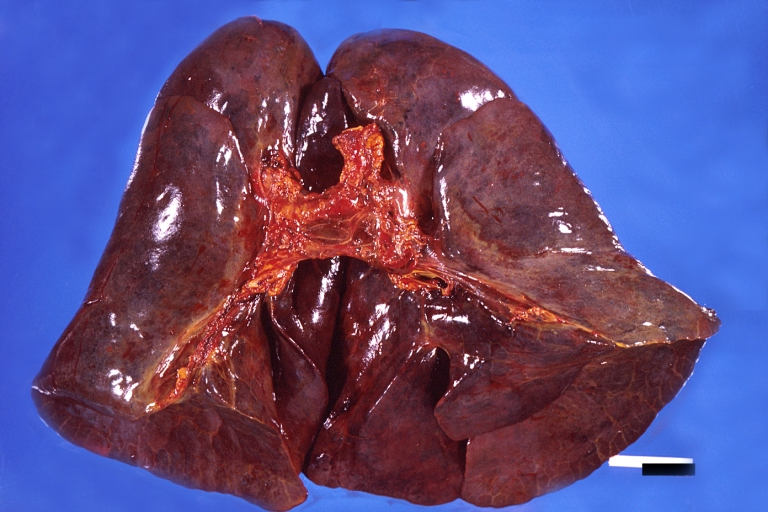
Lung, hemorrhagic bronchopneumonia, Wilson's disease
-
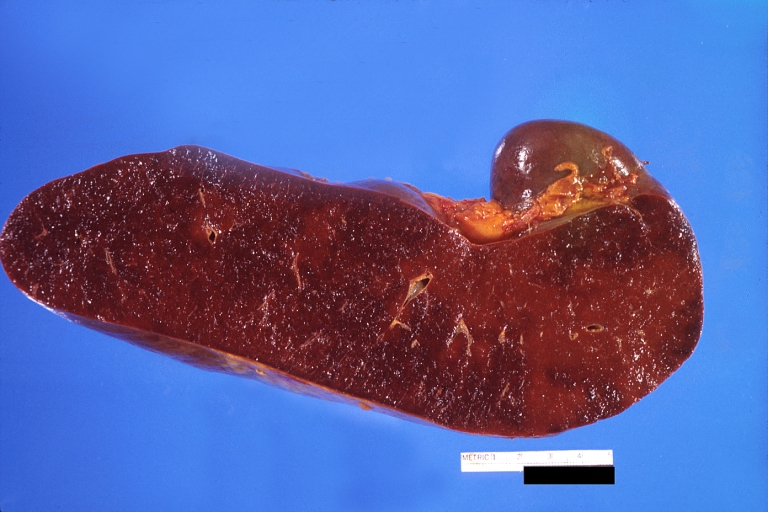
Spleen, congestion. Wilson's disease




{kind=link}
Microscopic pathology
- Histological examination of a liver biopsy may show the following:[10]
- Mild steatosis which is considered an early histological feature
- Glycogenated hepatic nuclei
- Hepatocellular necrosis
- Autoimmune hepatitis histologic features
- Fibrosis and cirrhosis (macronodular or micronodular) in advanced cases
- Fulminant liver falilure features which include:
- Hepatocellular degenration
- Parenchymal collapse
References
- ↑ Sandstead HH (1982). "Copper bioavailability and requirements". Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Sandstead HH (1982). "Copper bioavailability and requirements". Am J Clin Nutr. 35 (4): 809–14. PMID 6280488.
- ↑ Pfeiffer RF (2007). "Wilson's Disease". Semin Neurol. 27 (2): 123–32. doi:10.1055/s-2007-971173. PMID 17390257.
- ↑ Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997). "Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae". J Biol Chem. 272 (34): 21461–6. PMID 9261163.
- ↑ 5.0 5.1 5.2 5.3 de Bie P, Muller P, Wijmenga C, Klomp LW (2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". J. Med. Genet. 44 (11): 673–88. doi:10.1136/jmg.2007.052746. PMID 17717039. Unknown parameter
|month=ignored (help) - ↑ Grubenbecher S, Stüve O, Hefter H, Korth C (2006). "Prion protein gene codon 129 modulates clinical course of neurological Wilson disease". Neuroreport. 17 (5): 549–52. doi:10.1097/01.wnr.0000209006.48105.90. PMID 16543824.
- ↑ Sternlieb I, Twedt DC, Johnson GF; et al. (1977). "Inherited copper toxicity of the liver in Bedlington terriers". Proc. R. Soc. Med. 70 Suppl 3: 8–9. PMID 122681.
- ↑ van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002). "Identification of a new copper metabolism gene by positional cloning in a purebred dog population". Hum. Mol. Genet. 11 (2): 165–73. doi:10.1093/hmg/11.2.165. PMID 11809725.
- ↑ Müller T, van de Sluis B, Zhernakova A; et al. (2003). "The canine copper toxicosis gene MURR1 does not cause non-Wilsonian hepatic copper toxicosis". J. Hepatol. 38 (2): 164–8. doi:10.1016/S0168-8278(02)00356-2. PMID 12547404.
- ↑ Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW; et al. (2006). "MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation". AJNR Am J Neuroradiol. 27 (6): 1373–8. PMID 16775300.