Pheochromocytoma pathophysiology: Difference between revisions
| Line 7: | Line 7: | ||
==Pathophysiology== | ==Pathophysiology== | ||
Pheochromocytoma arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. | Pheochromocytoma arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. Traditionally pheochromocytoma known as the "10% tumor": | ||
Traditionally pheochromocytoma known as the "10% tumor": | |||
* Approximately 10% of patients have bilateral disease | * Approximately 10% of patients have bilateral disease | ||
* Approximately 10% of tumors are malignant | * Approximately 10% of tumors are malignant | ||
* Approximately 10% are located in chromaffin tissue outside of the adrenal gland | * Approximately 10% are located in chromaffin tissue outside of the adrenal gland, The most common extradrenal locations are the abdomen and thorax . | ||
* Approximately 10% occur in childhood | * Approximately 10% occur in childhood | ||
* Approximately 10% are familial | * Approximately 10% are familial | ||
| Line 20: | Line 18: | ||
Malignant and benign pheochromocytomas are the same; The only difference is the ability to spread locally and distant. <ref name="pmid10363888">{{cite journal| author=Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA et al.| title=Clinical experience over 48 years with pheochromocytoma. | journal=Ann Surg | year= 1999 | volume= 229 | issue= 6 | pages= 755-64; discussion 764-6 | pmid=10363888 | doi= | pmc=1420821 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=10363888 }}</ref> | Malignant and benign pheochromocytomas are the same; The only difference is the ability to spread locally and distant. <ref name="pmid10363888">{{cite journal| author=Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA et al.| title=Clinical experience over 48 years with pheochromocytoma. | journal=Ann Surg | year= 1999 | volume= 229 | issue= 6 | pages= 755-64; discussion 764-6 | pmid=10363888 | doi= | pmc=1420821 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=10363888 }}</ref> | ||
Pheochromocytomas can be familial and occur in patients with [[multiple endocrine neoplasia]] (MEN 2 and MEN 3). Patients with Von Hippel Lindau ([[VHL]]) may also develop pheocromocytoma.<ref name="pmid24642075">{{cite journal| author=Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM| title=The genetic basis of pheochromocytoma and paraganglioma: implications for management. | journal=Urology | year= 2014 | volume= 83 | issue= 6 | pages= 1225-32 | pmid=24642075 | doi=10.1016/j.urology.2014.01.007 | pmc=4572836 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=24642075 }}</ref> It is autosomal dominant inheritance and has two pathways of tumor pathogenesis. Cluster 1 tumorsare noradrenergic. Cluster 2 tumors are adrenergic.<ref name="pmid23933153">{{cite journal| author=King KS, Pacak K| title=Familial pheochromocytomas and paragangliomas. | journal=Mol Cell Endocrinol | year= 2014 | volume= 386 | issue= 1-2 | pages= 92-100 | pmid=23933153 | doi=10.1016/j.mce.2013.07.032 | pmc=3917973 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=23933153 }}</ref> | |||
●Cluster 1: | |||
•Succinate dehydrogenase (SDH) subunit genes | |||
•von Hippel-Lindau (VHL) disease | |||
•Fumarate hydratase gene mutations | |||
●Cluster 2: | |||
•Multiple endocrine neoplasia type 2A | |||
•Multiple endocrine neoplasia type 2B | |||
•Neurofibromatosis type 1 (NF1) | |||
==Gross Pathology== | ==Gross Pathology== | ||
A multinodular and multicentric pattern of growth of pheochromocytoma may be seen. | On gross pathology, A multinodular and multicentric pattern of growth of pheochromocytoma may be seen. | ||
<gallery> | <gallery> | ||
Image:Bilateral pheo MEN2.jpg|Bilateral pheochromocytoma in [[Multiple_endocrine_neoplasia_type_2|MEN2]]. Gross image. | Image:Bilateral pheo MEN2.jpg|Bilateral pheochromocytoma in [[Multiple_endocrine_neoplasia_type_2|MEN2]]. Gross image. | ||
| Line 29: | Line 43: | ||
==Microscopic Pathology== | ==Microscopic Pathology== | ||
Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma. | On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma. | ||
<gallery> | <gallery> | ||
Image:Adrenal pheochromocytoma (1) histopathology.jpg|[[Micrograph]] of pheochromocytoma. | Image:Adrenal pheochromocytoma (1) histopathology.jpg|[[Micrograph]] of pheochromocytoma. | ||
Revision as of 18:08, 30 June 2017
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma pathophysiology |
|
Risk calculators and risk factors for Pheochromocytoma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2]
Overview
On gross pathology, pheochromocytoma has a multinodular and a multicentric pattern of growth. On microscopic histopathological analysis, nesting (Zellballen) pattern composed of well-defined clusters of tumor cells separated by fibrovascular stroma is a characteristic finding.
Pathophysiology
Pheochromocytoma arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. Traditionally pheochromocytoma known as the "10% tumor":
- Approximately 10% of patients have bilateral disease
- Approximately 10% of tumors are malignant
- Approximately 10% are located in chromaffin tissue outside of the adrenal gland, The most common extradrenal locations are the abdomen and thorax .
- Approximately 10% occur in childhood
- Approximately 10% are familial
- Approximately 10% recur after being resected
- Approximately 10% of patients do not have hypertension
Malignant and benign pheochromocytomas are the same; The only difference is the ability to spread locally and distant. [1]
Pheochromocytomas can be familial and occur in patients with multiple endocrine neoplasia (MEN 2 and MEN 3). Patients with Von Hippel Lindau (VHL) may also develop pheocromocytoma.[2] It is autosomal dominant inheritance and has two pathways of tumor pathogenesis. Cluster 1 tumorsare noradrenergic. Cluster 2 tumors are adrenergic.[3]
●Cluster 1:
•Succinate dehydrogenase (SDH) subunit genes
•von Hippel-Lindau (VHL) disease
•Fumarate hydratase gene mutations
●Cluster 2:
•Multiple endocrine neoplasia type 2A
•Multiple endocrine neoplasia type 2B
•Neurofibromatosis type 1 (NF1)
Gross Pathology
On gross pathology, A multinodular and multicentric pattern of growth of pheochromocytoma may be seen.
-
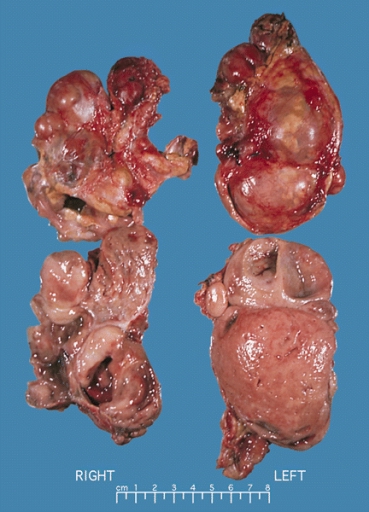
Bilateral pheochromocytoma in MEN2. Gross image.

Microscopic Pathology
On microscopic pathology, Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma.
-
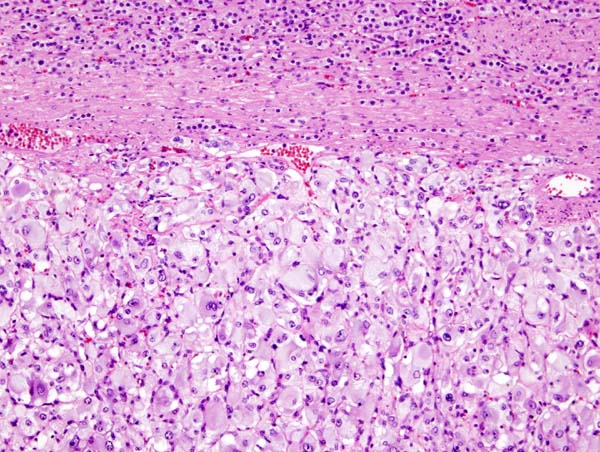
Micrograph of pheochromocytoma.
-
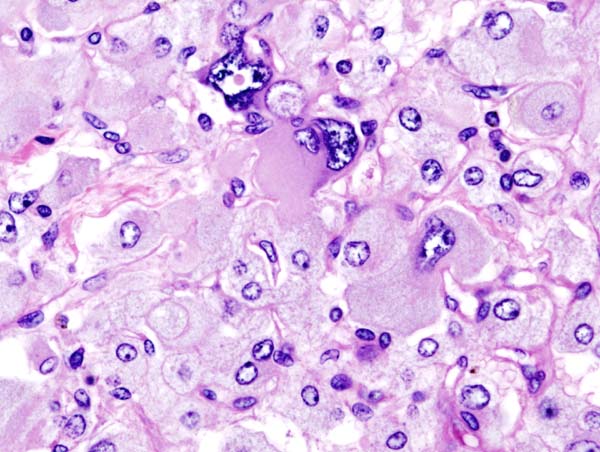
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen.
-
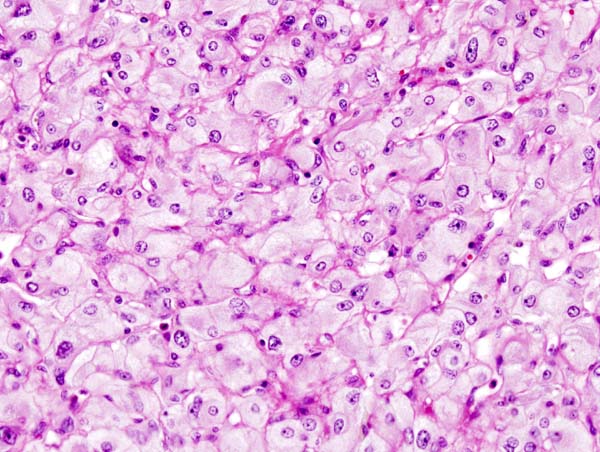
Micrograph of pheochromocytoma.
-
Micrograph of pheochromocytoma.
_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
Videos
{{#ev:youtube|7yjxG3KmX98}}
References
- ↑ Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA; et al. (1999). "Clinical experience over 48 years with pheochromocytoma". Ann Surg. 229 (6): 755–64, discussion 764-6. PMC 1420821. PMID 10363888.
- ↑ Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM (2014). "The genetic basis of pheochromocytoma and paraganglioma: implications for management". Urology. 83 (6): 1225–32. doi:10.1016/j.urology.2014.01.007. PMC 4572836. PMID 24642075.
- ↑ King KS, Pacak K (2014). "Familial pheochromocytomas and paragangliomas". Mol Cell Endocrinol. 386 (1–2): 92–100. doi:10.1016/j.mce.2013.07.032. PMC 3917973. PMID 23933153.