Pheochromocytoma pathophysiology: Difference between revisions
Irfan Dotani (talk | contribs) No edit summary |
|||
| Line 7: | Line 7: | ||
==Pathophysiology== | ==Pathophysiology== | ||
Mutations of the genes ''[[VHL]]'', ''RET'', ''NF1'', ''[[SDHB]]'', and ''[[SDHD]]'' are all known to cause familial pheochromocytoma | Pheochromocytoma arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. Mutations of the genes ''[[VHL]]'', ''RET'', ''NF1'', ''[[SDHB]]'', and ''[[SDHD]]'' are all known to cause familial pheochromocytoma. The most common extradrenal locations are the abdomen and thorax . | ||
Traditionally pheochromocytoma known as the "10% tumor": | Traditionally pheochromocytoma known as the "10% tumor": | ||
| Line 18: | Line 17: | ||
* Approximately 10% recur after being resected | * Approximately 10% recur after being resected | ||
* Approximately 10% of patients do not have [[hypertension]] | * Approximately 10% of patients do not have [[hypertension]] | ||
Malignant and benign pheochromocytomas are the same; The only difference is the ability to spread locally and distant. <ref name="pmid10363888">{{cite journal| author=Goldstein RE, O'Neill JA, Holcomb GW, Morgan WM, Neblett WW, Oates JA et al.| title=Clinical experience over 48 years with pheochromocytoma. | journal=Ann Surg | year= 1999 | volume= 229 | issue= 6 | pages= 755-64; discussion 764-6 | pmid=10363888 | doi= | pmc=1420821 | url=https://www.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&tool=sumsearch.org/cite&retmode=ref&cmd=prlinks&id=10363888 }}</ref> | |||
Pheochromocytoma can occur in patients with [[multiple endocrine neoplasia]] (MEN 2 and MEN 3). Patients with Von Hippel Lindau ([[VHL]]) may also develop pheocromocytoma. | Pheochromocytoma can occur in patients with [[multiple endocrine neoplasia]] (MEN 2 and MEN 3). Patients with Von Hippel Lindau ([[VHL]]) may also develop pheocromocytoma. | ||
Revision as of 17:41, 30 June 2017
|
Pheochromocytoma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Pheochromocytoma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Pheochromocytoma pathophysiology |
|
Risk calculators and risk factors for Pheochromocytoma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ahmad Al Maradni, M.D. [2]
Overview
On gross pathology, pheochromocytoma has a multinodular and a multicentric pattern of growth. On microscopic histopathological analysis, nesting (Zellballen) pattern composed of well-defined clusters of tumor cells separated by fibrovascular stroma is a characteristic finding.
Pathophysiology
Pheochromocytoma arise from chromaffin cells of the adrenal medulla and sympathetic ganglia. Mutations of the genes VHL, RET, NF1, SDHB, and SDHD are all known to cause familial pheochromocytoma. The most common extradrenal locations are the abdomen and thorax .
Traditionally pheochromocytoma known as the "10% tumor":
- Approximately 10% of patients have bilateral disease
- Approximately 10% of tumors are malignant
- Approximately 10% are located in chromaffin tissue outside of the adrenal gland
- Approximately 10% occur in childhood
- Approximately 10% are familial
- Approximately 10% recur after being resected
- Approximately 10% of patients do not have hypertension
Malignant and benign pheochromocytomas are the same; The only difference is the ability to spread locally and distant. [1]
Pheochromocytoma can occur in patients with multiple endocrine neoplasia (MEN 2 and MEN 3). Patients with Von Hippel Lindau (VHL) may also develop pheocromocytoma.
Gross Pathology
A multinodular and multicentric pattern of growth of pheochromocytoma may be seen.
-
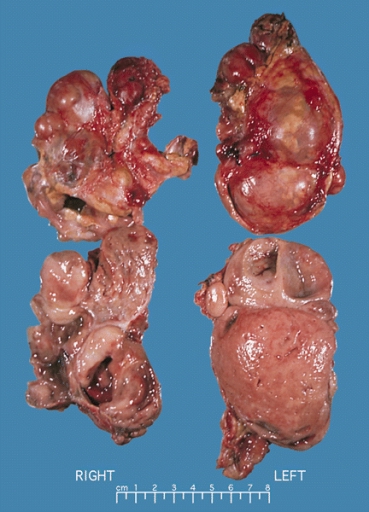
Bilateral pheochromocytoma in MEN2. Gross image.

Microscopic Pathology
Pheochromocytoma typically demonstrates a nesting (Zellballen) pattern on microscopy. This pattern is composed of well-defined clusters of tumor cells containing eosinophilic cytoplasm separated by fibrovascular stroma.
-
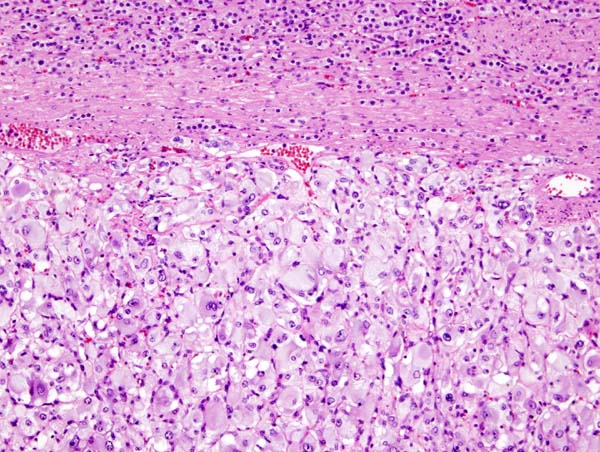
Micrograph of pheochromocytoma.
-
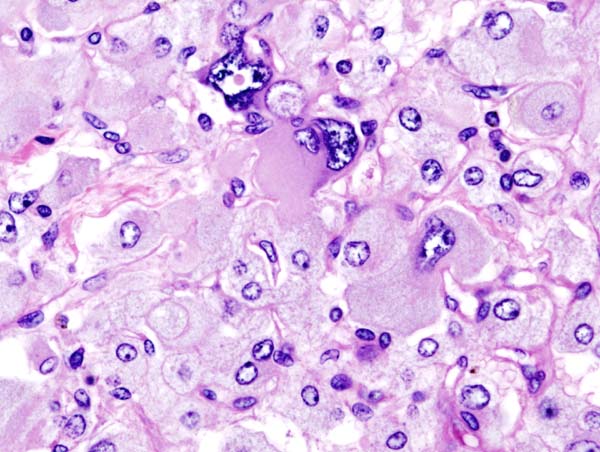
Histopathology of adrenal pheochromocytoma. Adrenectomy specimen.
-
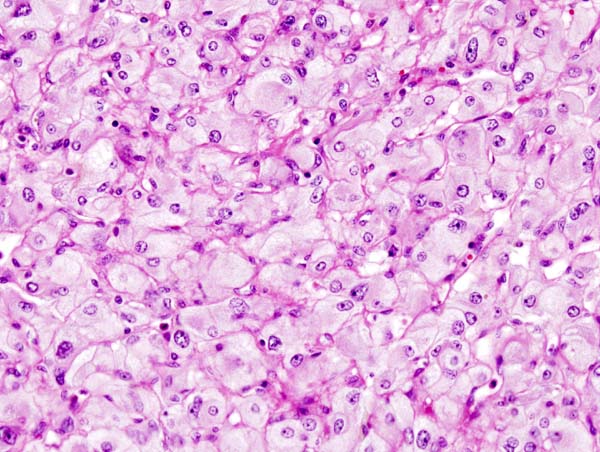
Micrograph of pheochromocytoma.
-
Micrograph of pheochromocytoma.
_histopathology.jpg)
_histopathology.jpg)
_histopathology.jpg)
Videos
{{#ev:youtube|7yjxG3KmX98}}