Achondroplasia: Difference between revisions
| Line 204: | Line 204: | ||
File:Achondroplasia (7).jpg | File:Achondroplasia (7).jpg | ||
File:Achondroplasia (6).jpg | File:Achondroplasia (6).jpg | ||
File:Achondroplasia (9).jpg | |||
File:Achondroplasia (10).jpg | |||
</gallery> | </gallery> | ||
Revision as of 12:08, 30 March 2019

| https://https://www.youtube.com/watch?v=gt-SAjuikLM%7C350}} |
Template:DiseaseDisorder infobox Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Mohammadmain Rezazadehsaatlou[2].
|
WikiDoc Resources for Achondroplasia |
|
Articles |
|---|
|
Most recent articles on Achondroplasia Most cited articles on Achondroplasia |
|
Media |
|
Powerpoint slides on Achondroplasia |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Achondroplasia at Clinical Trials.gov Trial results on Achondroplasia Clinical Trials on Achondroplasia at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Achondroplasia NICE Guidance on Achondroplasia
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Achondroplasia Discussion groups on Achondroplasia Patient Handouts on Achondroplasia Directions to Hospitals Treating Achondroplasia Risk calculators and risk factors for Achondroplasia
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Achondroplasia |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Synonyms and Keywords: ACH; Achondroplastic dwarfism
Overview
Achondroplasia is known as one of the most common forms of skeletal dysplasia that causes dwarfism. It is a rare genetic disorder characterized by specific features such as:
- Short stature (usually shorter than 4 feet 6 inches)
- An unusually large head (macrocephaly) with a notable forehead (frontal bossing) and flat (depressed) nasal bridge;
- Short arms
- Short legs
- Short hands with fingers that represent a “trident” or three-pronged position during extension
- Prominent abdomen (due to specific curve of the spine)
- Prominent buttocks (due to inward curve of the spine)
Achondroplasia occurs due to a specific changes (mutations) of a gene known as fibroblast growth factor receptor 3 (FGFR3).
Achondroplasia appears to affect males and females equally. The estimated frequency of achondroplasia has ranged from about one in 15,000 to one in 35,000 births.
The primary problem in patients with achondroplasia is abnormal endochondral ossification while the Periosteal and intramembranous ossification is normal. Tubular bones are short and broad, reflecting normal periosteal growth. The iliac crest apophyses (appositional growth) are normal. But the growth of the triradiate cartilage (endochondral growth) is not normal. Consequently, the patterns of defect help to explain the clinical and radiographic characteristics of achondroplasia in this patients.
Historical Perspective
The noun dwarf stems from Old English dweorg, originally referring to a being from Germanic mythology—a dwarf—that dwells in mountains and in the earth, and is associated with wisdom, smithing, mining, and crafting. Nowadays. the terms "dwarf", "little person", "LP", and "person of short stature" are now generally considered acceptable by most people affected by these disorders. However, the plural "dwarfs" as opposed to "dwarves" is generally preferred in the medical context, possibly because the plural "dwarves" was popularized by author J.R.R. Tolkien, describing a race of characters in his The Lord of the Rings books resembling Norse dwarfs. "Midget", whose etymology indicates a "tiny biting insect", came into prominence in the mid-19th century after Harriet Beecher Stowe used it in her novels Sunny Memories of Foreign Lands and Old Town Folks where she described children and an extremely short man, respectively. Later some people of short stature considered the word to be offensive because it was the descriptive term applied to P. T. Barnum's dwarfs used for public amusement during the freak show era. It is also not considered accurate as it is not a medical term or diagnosis, though it is sometimes used as a slang term to describe those who are particularly short, whether or not they have dwarfism. Meanwhile, the specific term for describing a person with notable shorter stature has historically been ambiguous, and has developed euphemistically over the past centuries.
In art, literature, and movies, dwarfs are rarely depicted as ordinary people who are very short but rather as a species apart. Artistic representations of dwarfism can be found on Greek vases and other ancient artifacts, including ancient Egyptian art in which dwarfs are likely to have been seen as a divine manifestation, with records indicating they could reach high positions in society.
- 3000 BC in ancient Egypt: The typical depiction of a dwarf in Egyptian art is pragmatic because we can found a normally grown torso and head, but visibly shortened and slightly bent arms and legs. Those similarity point to achondroplasia and hypochondroplasia as the conditions responsible for the dwarfism of the individual.

Although this term is inaccurate from a histopathologic view but it use is universally accepted by the International Working Group on Constitutional Diseases of the Bone.
Classification
Disorders that cause Achondroplasia can be categorized according to various names, which are usually permutations of the following roots:
- Location based
- rhizomelic = root, i.e., bones of the upper arm or thigh
- mesomelic = middle, i.e., bones of the forearm or lower leg
- acromelic = end, i.e., bones of hands and feet.
- micromelic = entire limbs are shortened
- Source Based
- chondro = of cartilage
- osteo = of bone
- spondylo = of the vertebrae
- plasia = form
- trophy = growth
Pathophysiology
Conditions leading to the Achondroplasia are frequently referred to as short-limb or short-trunk types considering which the trunk or limbs are mostly involved. Achondroplasia, hypochondroplasia, and metaphyseal chondrodysplasias are considered short-limb dwarfing conditions. Normally, these patients' sitting height is within normal range. Additional terms used to describe the segment of the limb with the greatest involvement include the following:
- Rhizomelic (proximal)
- Mesomelic (middle)
- Acromelic (distal)
Causes
Genetic pathway
A single gene responsible to the short arm is from the chromosome 4 (band 4p16.3) is transmitted as an autosomal dominant trait. Around 80% of cases result from a new onset mutation. Most parents are of average size and have no family history of a dwarfing condition. The risk of the parents producing a second affected child is almost insignificant. Reports have estimated that there is a 1 in 443 risk of recurrence of achondroplasia in the siblings of an affected child with unaffected parents. This is because of gonadal mosaicism in the parents. Average-sized siblings have no increased risk of producing a child with achondroplasia. When one parent has achondroplasia, the chance of transmitting this gene to each child is 50%; while, in both parents wit achondroplasia, 50% of their offspring are heterozygous and affected, 25% are homozygous, which is ordinarily fatal in the first few months of life, and 25% are unaffected. .
Molecular pathway
Fibroblast growth factors (FGFs) are structurally related proteins associated with cell growth, migration, wound healing, and angiogenesis. At the cellular level, their function is mediated by transmembrane tyrosine kinase receptors, known as FGF receptors (FGFRs).
Mutation in FGFR3 is responsible for achondroplasia, hypochondroplasia, and thanatophoric dysplasia. The primary function of FGFR3 is to limit osteogenesis. Mutation causes enhancement in its function of limiting endochondral ossification. Mutation in FGFR3 in achondroplasia is due to transition of guanine to adenine (G to A) at nucleotide 1138 of complimentary DNA.
Associated syndromes
Up to this time there are only two reports regarding the achondroplasia associated with Down syndrome. On the basis of the current birth rate, the calculated risk of association is 1 case in every 8 years in the United States of america.
Differentiating Achondroplasia from Other Diseases
- Thanatophoric dwarfism
- Achondrogenesis
- Chondroectodermal dysplasia ( Ellis-van Creveld syndrome )
- Metatrophic dwarfism
- Asphyxiating thoracic dysplasia
- Chondrodysplasia punctata (Conradi disease)
- Pseudoachondroplastic dysplasia
- Metaphyseal chondrodysplasia (Schmid type)
- Diastrophic Dysplasia
- Spondyloepiphyseal Dysplasia
Epidemiology and Demographics
Worldwide, achondroplasia is the most common skeletal dysplasia, affecting about 1 in every 40,000 children. (This number varies, depending on the source.) About 80% of all "little people" have achondroplasia. Approximately 150,000 persons have achondroplasia worldwide. The worldwide population of little people is approximately 190,000. achondroplasiaoccurs with equal frequency in males and females. (It is inherited in an autosomal dominant manner.) It occurs in all the races with equal frequency. Its been estimated that around 10,000 individuals to have achondroplasia in the United States.
Risk Factors
Screening
As the first step:In parents with normal stature, the diagnosis may only be suspected on the basis of the observation of disproportionately short limbs in the fetus upon ultrasonographic evaluation; while one or both parents have this condition the diagnosis can be made accordingly. But it should be noted that the in most cases, the specific diagnosis cannot be made with certainty until birth. Caution should be exercised in counseling the family.
Antenatally sonographic features include:
- short femur length measurement: often well below the 5th centile
- the femur length (FL) to biparietal diameter (BPD)
- trident hand: 2,3 and 4 fingers appearing separated and similar in length
- separation of 1st, 2nd, 3rd and 4th fingers
- protruding forehead: frontal bossing
- depressed nasal bridge
The diagnosis should be confirmed at birth by means of radiographic studies. The measurements, including arm span, occipital frontal circumference, body length, and ratio of upper body to lower body, should be documented.
As the second step: Plasma can be analyzed for the FGFR3 mutation in suspected mother when a short-limb skeletal dysplasia is diagnosed antenatally by means of ultrasonography evaluations. DNA testing can be performed when both of the parents are affected. Infants with affected genes from both the parents (double homozygous) are either stillborn or die shortly after birth. This can be confirmatory for achondroplasia and can help the family to make educated decisions.
Natural History, Complications, and Prognosis
Natural History
In children younger than 4 years, mortality related to the consequence of brainstem compression, which causes sudden death. In individuals aged 5-24 years, central nervous system (CNS) and respiratory abnormalities are the common causes of death. In patients aged 25-54 years, cardiovascular problems are the most frequent causes of death.
Morbidity associated with achondroplasia may include the following:
- Recurrent otitis media
- Hearing loss
- Neurologic complications due to cervicomedullary compression (eg, hypotonia, respiratory insufficiency, apnea, cyanotic episodes, feeding problems, quadriparesis)
- Obstructive and restrictive respiratory complications (eg, upper airway obstruction, pneumonia, apnea)
- Hydrocephalus
- Spinal deformities (eg, kyphosis, lordosis, scoliosis)
- Obesity
- Spinal canal stenosis
- Genu varum
- Cardiovascular complications
Complications
Prognosis
Treatment
Pathophysiology
Achondroplasia is a result of an autosomal dominant mutation in the fibroblast growth factor receptor gene 3 (FGFR3), which causes an abnormality of cartilage formation. FGFR3 normally has a negative regulatory effect on bone growth. In achondroplasia, the mutated form of the receptor is constitutively active and this leads to severely shortened bones.
People with achondroplasia have one normal copy of the fibroblast growth factor receptor 3 gene and one mutant copy. Two copies of the mutant gene are invariably fatal before, or shortly after birth. Only one copy of the gene needs to be present for the disorder to occur. Therefore, a person with achondroplasia has a 50% chance of passing on the gene to their offspring, meaning that there will be a 50% chance that each child will have achondroplasia. Since two copies (Homozygous) are fatal, if two people with achondroplasia have a child, there is a 25% chance of the child dying shortly after birth, a 50% chance the child will have achondroplasia, and a 25% chance the child will have a normal phenotype. However, in 3 out of 4 cases, people with achondroplasia are born to parents who don't have the condition. This is the result of a new mutation.
New gene mutations are associated with increasing paternal age (over 35 years). Studies have demonstrated that new gene mutations are exclusively inherited from the father and occur during spermatogenesis (as opposed to resulting from a gonadal mosaicism). More than 99% of achondroplasia is caused by two different mutations in the fibroblast growth factor receptor 3 (FGFR3). In about 98% of cases, a G to A point mutation at nucleotide 1138 of the FGFR3 gene causes a glycine to arginine substitution (Bellus et al 1995, Shiang et al 1994, Rousseau et al 1996). About 1% of cases are caused by a G to C point mutation at nucleotide 1138.
There are two other syndromes with a genetic basis similar to achondroplasia: hypochondroplasia and thanatophoric dysplasia. Both of these disorders are also caused by a genetic mutation in the FGFR3 gene.
Diagnosis
Symptoms
- Frequent ear infections (due to Eustachian tube blockages)
- Sleep apnea (which can be central or obstructive)
Physical Examination
General
- Motor milestones may lag by 3-6 months
- Onset and frequent otitis media
- Conductive hearing loss
- Upper-airway obstruction, small chest wall, pectus excavatum
- Delayed gross motor development
- Achondroplastic dwarfs have short stature , with an average adult height of 131 cm (4 feet 3.8 inches) for males and 123 cm (4 feet 0.6 inches) for females.
- Stenosis of the spinal canal and intervertebral foramen
- low back pain, leg pain, dysesthesia, paresthesia, paraparesis, incontinence, and neurogenic claudication.
- Spinal kyphosis (convex curvature of the spine) or lordosis (concave curvature of the spine)
HEENT
- Macrocephaly
- Hydrocephalus may be present
- A large head with prominent forehead
- Small midface or midface hypoplasia
- Flattened nasal bridge
Extremities
- Joint laxity
- Shortening of the proximal limbs (termed rhizomelic shortening)
- Short fingers and toes
- Varus (bowleg) or valgus (knock knee) deformities
- Fibromyalgia (trigger points located in the lower part of the back)
- trochanteric bursitis
Imaging Studies
Radiography
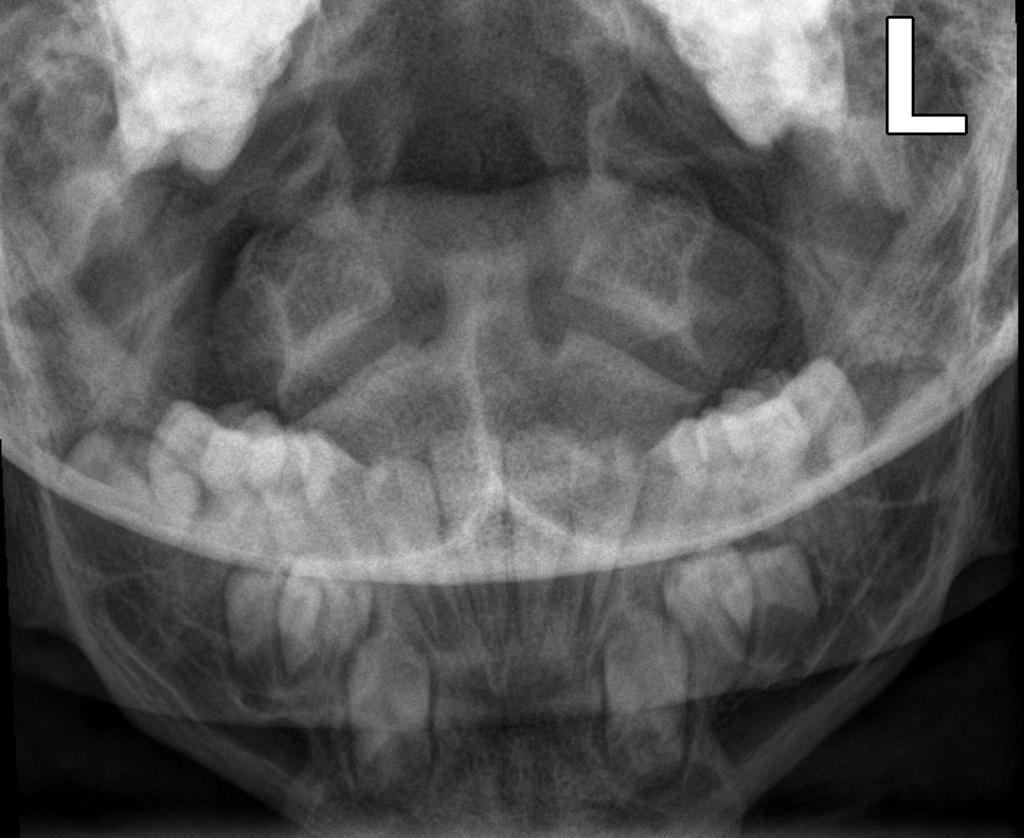
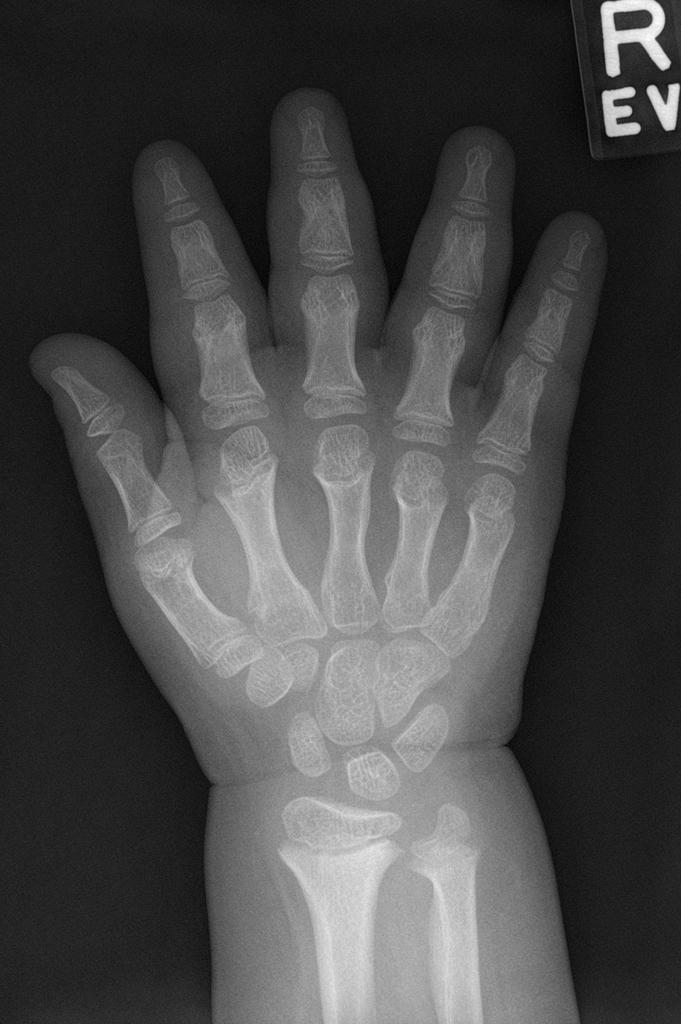
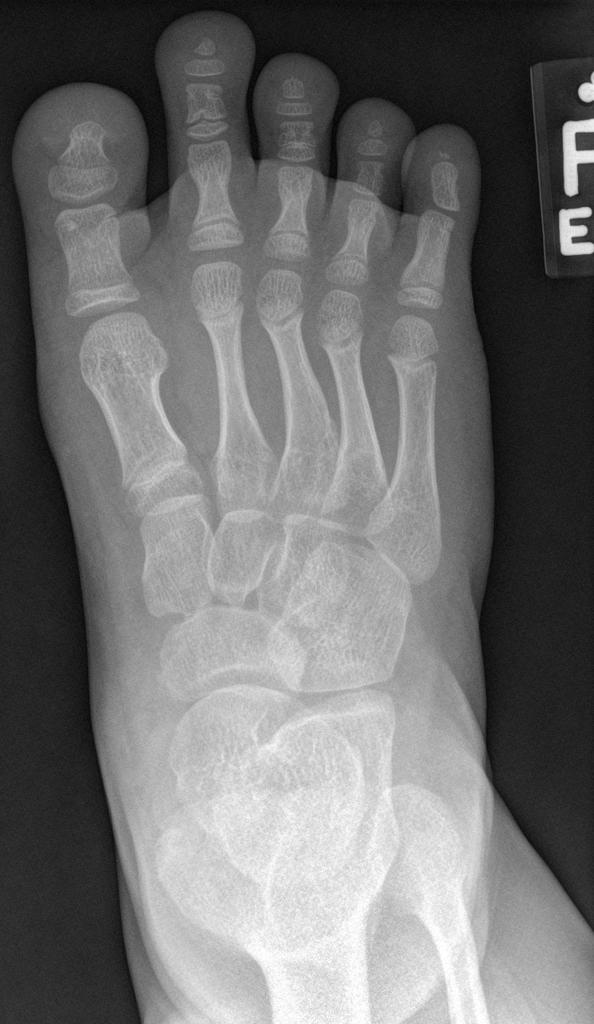
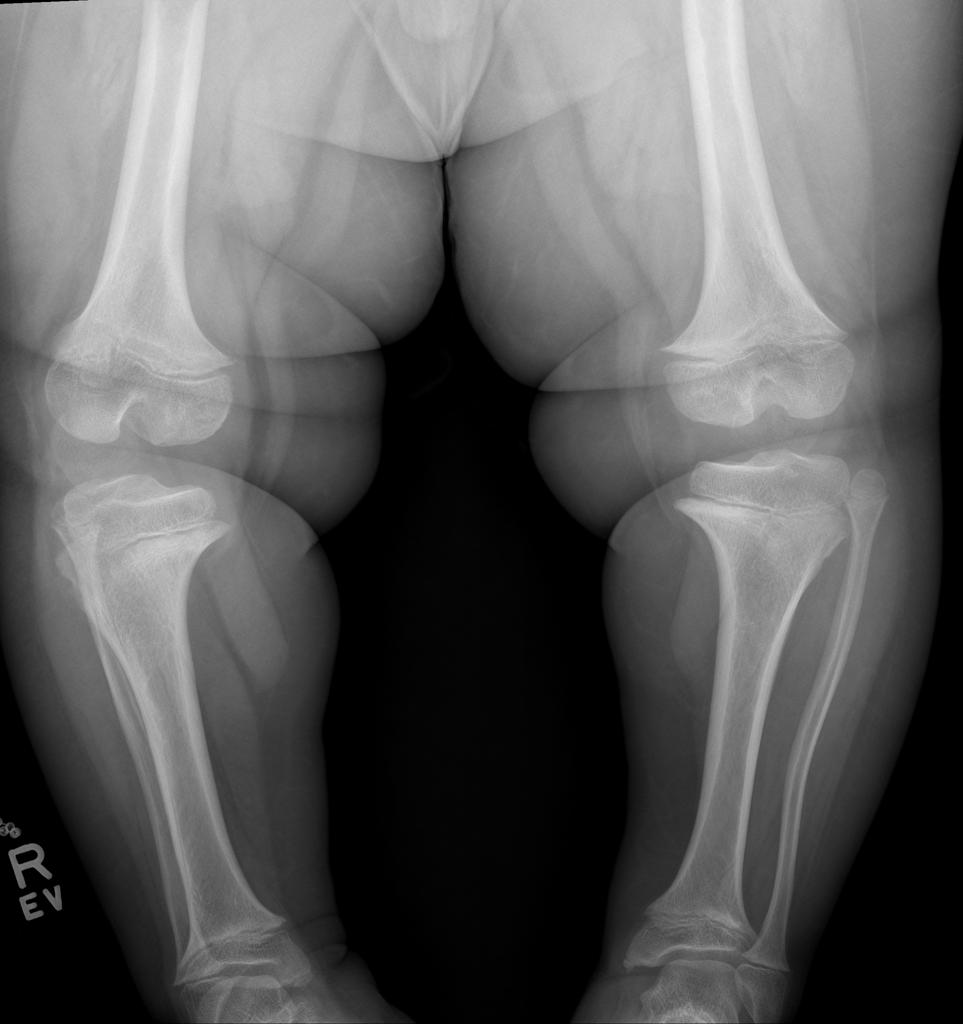
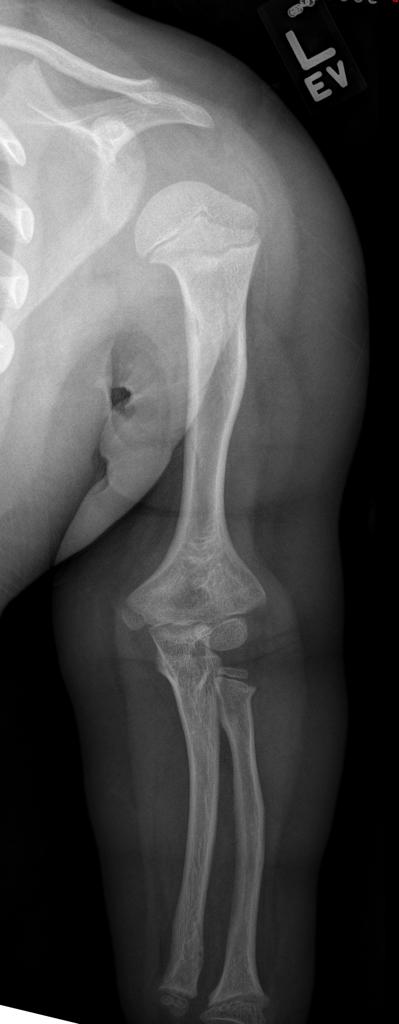
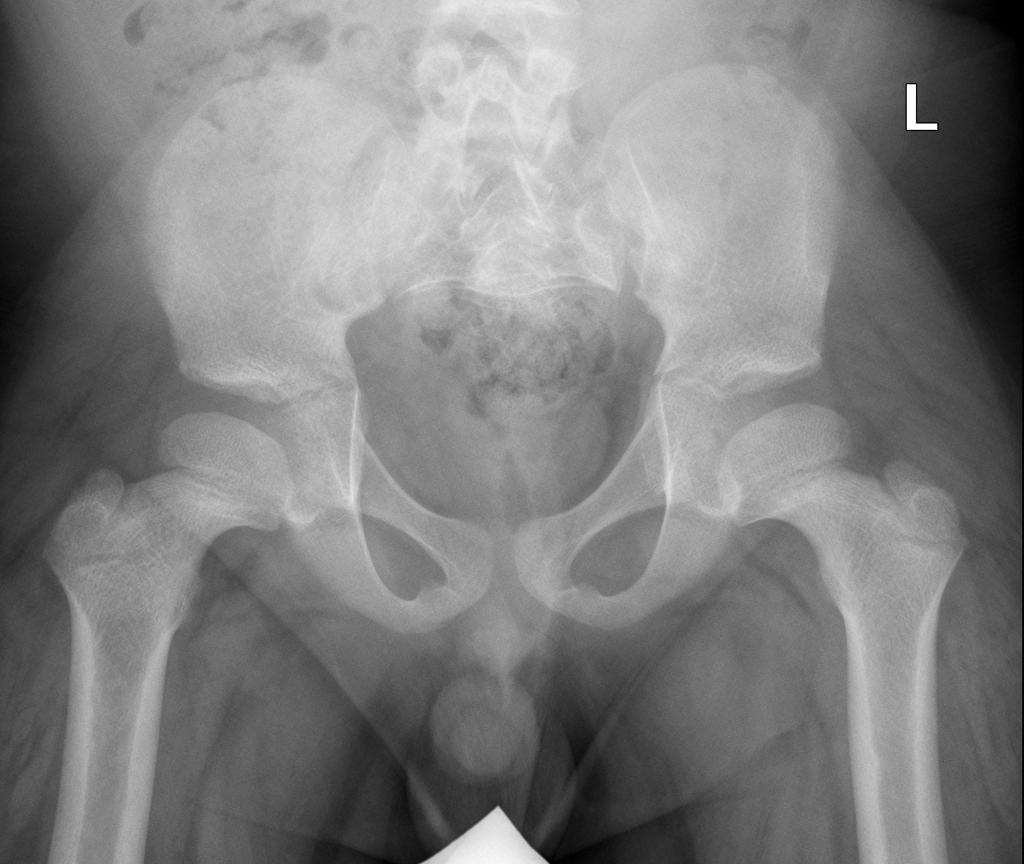
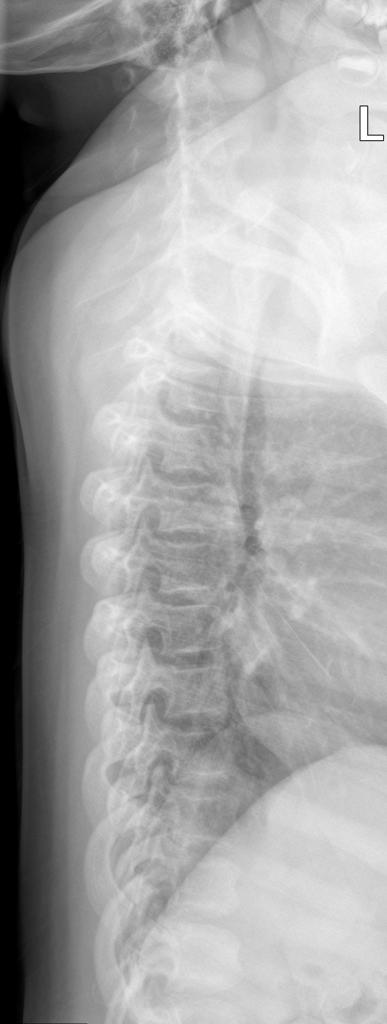
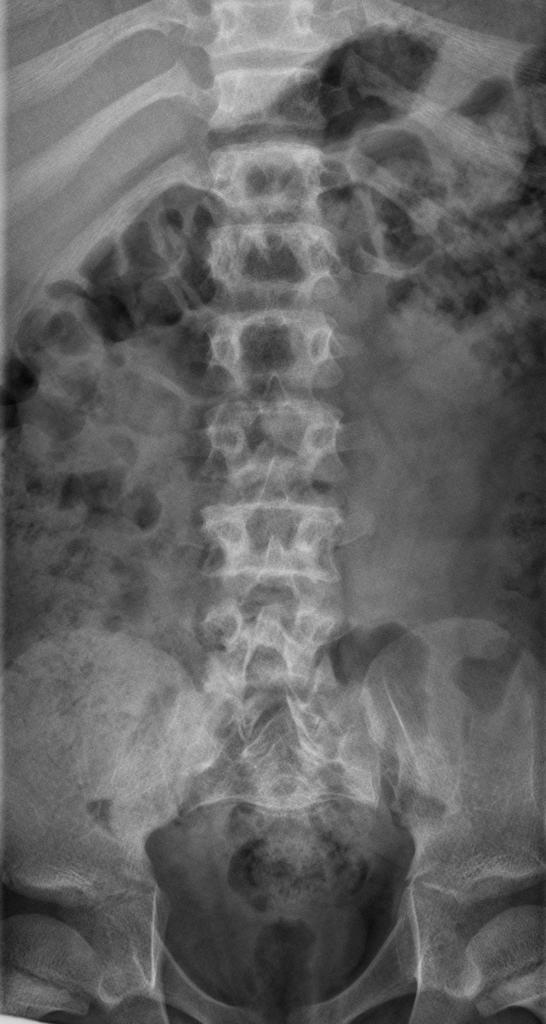
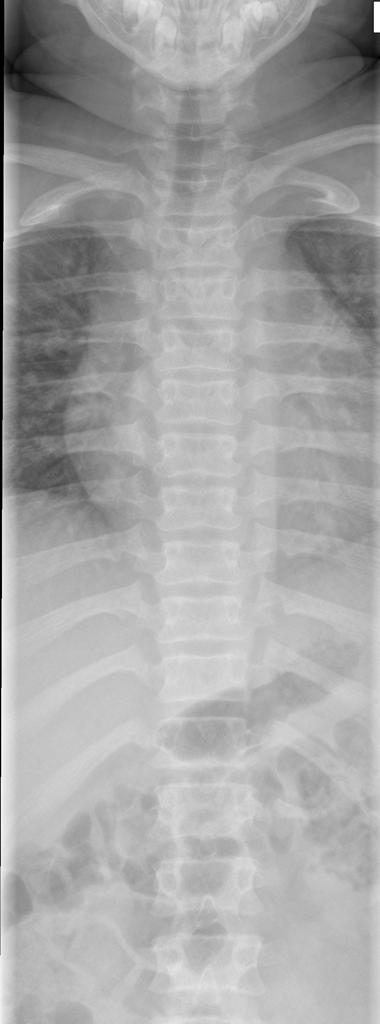
A skeletal survey is useful to confirm the diagnosis of achondroplasia. Radiographs of the skull, spine, and extremities show the specific features. A lateral skull radiograph demonstrates midface hypoplasia, enlarged calvaria, frontal prominence, and shortening of the base of the skull. Skull films demonstrate a large skull with a narrow foramen magnum, and relatively small skull base. The vertabral bodies are short and flattened with relatively large intervertebral disk height, and there is congenitally narrowed spinal canal. The iliac wingsare small and squared, with a narrow sciatic notch and horizontal acetabular roof. The tubular bones are short and thick with metaphyseal cupping and flaring and irregular growth plates. Fibular overgrowth is present. The hand is broad with short metacarpals and phalanges, and a trident configuration. The ribs are short with cupped anterior ends. If the radiographic features are not classic, a search for a different diagnosis should be entertained. Because of the extremely deformed bone structure, people with achondroplasia are often double jointed.
More obvious signs are a prominent forehead, flat nose bridge, protruding jaw, and crowded teeth.
- Shortening of tubular bones with a normal shaft caliber
- Short phalanges
- Squared iliac wings and narrow sacroiliac notch: champagne glass
- Fingers widely opposed and equal length: trident hands
- Enlarged skull vault and mandible
- Small foramen magnum
- Decreased lumber interpediculate distance and narrow spinal canal
.
-
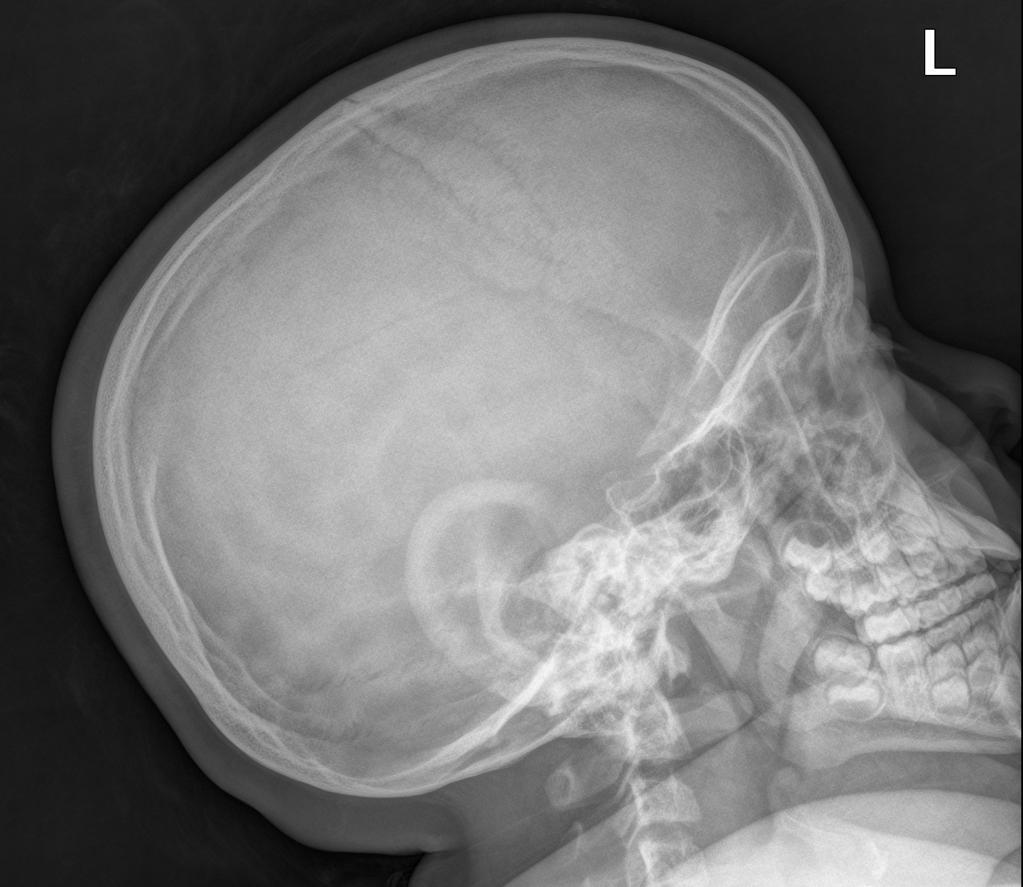
Case courtesy of Dr Hani Salam, <a href="https://radiopaedia.org/">Radiopaedia.org</a>. From the case <a href="https://radiopaedia.org/cases/9294">rID: 9294</a>
-
-
-
-
-
-
-
-
-

.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
The diagnosis can be made by fetal ultrasound by progressive discordance between the femur length and biparietal diameter by age. The trident hand configuration can be seen if the fingers are fully extended.
Treatment
At present, there is no actual treatment for achondroplasia.
Although used by those without achondroplasia to aid in growth, growth hormone won't help people with achondroplasia. However, if desired, the controversial surgery (among dwarfs) of limb-lengthening will lengthen the legs and arms of someone with achondroplasia.
References
de:Achondroplasie fa:آکندروپلازی gl:Acondroplasia ko:연골무형성증 nl:Achondroplasie sr:Ахондроплазија fi:Akondroplasia