Achondroplasia

| https://https://www.youtube.com/watch?v=gt-SAjuikLM%7C350}} |
Template:DiseaseDisorder infobox Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Mohammadmain Rezazadehsaatlou[2].
|
WikiDoc Resources for Achondroplasia |
|
Articles |
|---|
|
Most recent articles on Achondroplasia Most cited articles on Achondroplasia |
|
Media |
|
Powerpoint slides on Achondroplasia |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Achondroplasia at Clinical Trials.gov Trial results on Achondroplasia Clinical Trials on Achondroplasia at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Achondroplasia NICE Guidance on Achondroplasia
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Achondroplasia Discussion groups on Achondroplasia Patient Handouts on Achondroplasia Directions to Hospitals Treating Achondroplasia Risk calculators and risk factors for Achondroplasia
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Achondroplasia |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Synonyms and Keywords: ACH; Achondroplastic dwarfism, dwarf, achondroplastic
Achondroplasia is known as one of the most common forms of skeletal dysplasia that causes dwarfism[5]. It is a rare genetic disorder characterized by specific features such as[6][7]:
- Short stature (usually shorter than 4 feet 6 inches)
- An unusually large head (macrocephaly) with a notable forehead (frontal bossing) and flat (depressed) nasal bridge;
- Short arms
- Short legs
- Short hands with fingers that represent a “trident” or three-pronged position during extension
- Prominent abdomen (due to specific curve of the spine)
- Prominent buttocks (due to inward curve of the spine)
Achondroplasia occurs due to a specific changes (mutations) of a gene known as fibroblast growth factor receptor 3 (FGFR3)[8].
Achondroplasia appears to affect males and females equally. The estimated frequency of achondroplasia has ranged from about one in 15,000 to one in 35,000 births[9].
The primary problem in patients with achondroplasia is abnormal endochondral ossification while the Periosteal and intramembranous ossification is normal. Tubular bones are short and broad, reflecting normal periosteal growth. The iliac crest apophyses (appositional growth) are normal. But the growth of the triradiate cartilage (endochondral growth) is not normal. Consequently, the patterns of defect help to explain the clinical and radiographic characteristics of achondroplasia in this patients[8].
The noun dwarf stems from Old English dweorg, originally referring to a being from Germanic mythology—a dwarf—that dwells in mountains and in the earth, and is associated with wisdom, smithing, mining, and crafting. Nowadays. the terms "dwarf", "little person", "LP", and "person of short stature" are now generally considered acceptable by most people affected by these disorders. However, the plural "dwarfs" as opposed to "dwarves" is generally preferred in the medical context, possibly because the plural "dwarves" was popularized by author J.R.R. Tolkien, describing a race of characters in his The Lord of the Rings books resembling Norse dwarfs. "Midget", whose etymology indicates a "tiny biting insect", came into prominence in the mid-19th century after Harriet Beecher Stowe used it in her novels Sunny Memories of Foreign Lands and Old Town Folks where she described children and an extremely short man, respectively. Later some people of short stature considered the word to be offensive because it was the descriptive term applied to P. T. Barnum's dwarfs used for public amusement during the freak show era. It is also not considered accurate as it is not a medical term or diagnosis, though it is sometimes used as a slang term to describe those who are particularly short, whether or not they have dwarfism. Meanwhile, the specific term for describing a person with notable shorter stature has historically been ambiguous, and has developed euphemistically over the past centuries[9].
In art, literature, and movies, dwarfs are rarely depicted as ordinary people who are very short but rather as a species apart. Artistic representations of dwarfism can be found on Greek vases and other ancient artifacts, including ancient Egyptian art in which dwarfs are likely to have been seen as a divine manifestation, with records indicating they could reach high positions in society[15].
- 3000 BC in ancient Egypt: The typical depiction of a dwarf in Egyptian art is pragmatic because we can found a normally grown torso and head, but visibly shortened and slightly bent arms and legs. Those similarity point to achondroplasia and hypochondroplasia as the conditions responsible for the dwarfism of the individual.

Although this term is inaccurate from a histopathologic view but it use is universally accepted by the International Working Group on Constitutional Diseases of the Bone[9].
Disorders that cause Achondroplasia can be categorized according to various names, which are usually permutations of the following roots[9]:
- Location based
- rhizomelic = root, i.e., bones of the upper arm or thigh
- mesomelic = middle, i.e., bones of the forearm or lower leg
- acromelic = end, i.e., bones of hands and feet.
- micromelic = entire limbs are shortened
- Source Based
- chondro = of cartilage
- osteo = of bone
- spondylo = of the vertebrae
- plasia = form
- trophy = growth
Pathophysiology[16]
Conditions leading to the Achondroplasia are frequently referred to as short-limb or short-trunk types considering which the trunk or limbs are mostly involved. Achondroplasia, hypochondroplasia, and metaphyseal chondrodysplasias are considered short-limb dwarfing conditions. Normally, these patients' sitting height is within normal range. Additional terms used to describe the segment of the limb with the greatest involvement include the following:
- Rhizomelic (proximal)
- Mesomelic (middle)
- Acromelic (distal)
Causes [15]
Genetic pathway
A single gene responsible to the short arm is from the chromosome 4 (band 4p16.3) is transmitted as an autosomal dominant trait. Around 80% of cases result from a new onset mutation. Most parents are of average size and have no family history of a dwarfing condition. The risk of the parents producing a second affected child is almost insignificant. Reports have estimated that there is a 1 in 443 risk of recurrence of achondroplasia in the siblings of an affected child with unaffected parents. This is because of gonadal mosaicism in the parents. Average-sized siblings have no increased risk of producing a child with achondroplasia. When one parent has achondroplasia, the chance of transmitting this gene to each child is 50%; while, in both parents wit achondroplasia, 50% of their offspring are heterozygous and affected, 25% are homozygous, which is ordinarily fatal in the first few months of life, and 25% are unaffected [10].
Molecular pathway
Fibroblast growth factors (FGFs) are structurally related proteins associated with cell growth, migration, wound healing, and angiogenesis. At the cellular level, their function is mediated by transmembrane tyrosine kinase receptors, known as FGF receptors (FGFRs)[10][17].
Mutation in FGFR3 is responsible for achondroplasia, hypochondroplasia, and thanatophoric dysplasia. The primary function of FGFR3 is to limit osteogenesis. Mutation causes enhancement in its function of limiting endochondral ossification. Mutation in FGFR3 in achondroplasia is due to transition of guanine to adenine (G to A) at nucleotide 1138 of complimentary DNA.
Associated syndromes
Up to this time there are only two reports regarding the achondroplasia associated with Down syndrome. On the basis of the current birth rate, the calculated risk of association is 1 case in every 8 years in the United States of america [18].
Differentiating Achondroplasia from Other Diseases [19]
- Thanatophoric dwarfism
- Achondrogenesis
- Chondroectodermal dysplasia ( Ellis-van Creveld syndrome )
- Metatrophic dwarfism
- Asphyxiating thoracic dysplasia
- Chondrodysplasia punctata (Conradi disease)
- Pseudoachondroplastic dysplasia
- Metaphyseal chondrodysplasia (Schmid type)
- Diastrophic Dysplasia
- Spondyloepiphyseal Dysplasia
Epidemiology and Demographics
Worldwide, achondroplasia is known as the most common skeletal dysplasia, affecting about 1 in every 40,000 children. (This number varies, depending on the source.) About 80% of all "little people" have achondroplasia. Approximately 150,000 persons have achondroplasia worldwide. The worldwide population of little people is approximately 190,000. achondroplasiaoccurs with equal frequency in males and females. (It is inherited in an autosomal dominant manner.) It occurs in all the races with equal frequency. Its been estimated that around 10,000 individuals to have achondroplasia in the United States.
No documented race predilection is noted in this regard[20].
Risk Factors
The risk factors of Achondroplasia include[20]:
- In 10% of Achondroplasia cases: the condition is inherited as an autosomal dominant trait, i.e. the FGFR3 gene mutation is passed on to the child from an affected parent. Thus, a positive family history of the condition increases related risk.
- In 90% of Achondroplasia cases: the FGFR3 gene mutation is a new mutation (whose parents do not have the mutated gene). The risk factors in these individuals remain unknown.
Screening
As the first step:In parents with normal stature, the diagnosis may only be suspected on the basis of the observation of disproportionately short limbs in the fetus upon ultrasonographic evaluation; while one or both parents have this condition the diagnosis can be made accordingly. But it should be noted that the in most cases, the specific diagnosis cannot be made with certainty until birth. Caution should be exercised in counseling the family.
Antenatally sonographic features include:
- short femur length measurement: often well below the 5th centile
- the femur length (FL) to biparietal diameter (BPD)
- trident hand: 2,3 and 4 fingers appearing separated and similar in length
- separation of 1st, 2nd, 3rd and 4th fingers
- protruding forehead: frontal bossing
- depressed nasal bridge
The diagnosis should be confirmed at birth by means of radiographic studies. The measurements, including arm span, occipital frontal circumference, body length, and ratio of upper body to lower body, should be documented.
As the second step: Plasma can be analyzed for the FGFR3 mutation in suspected mother when a short-limb skeletal dysplasia is diagnosed antenatally by means of ultrasonography evaluations. DNA testing can be performed when both of the parents are affected. Infants with affected genes from both the parents (double homozygous) are either stillborn or die shortly after birth. This can be confirmatory for achondroplasia and can help the family to make educated decisions.
Natural History[16]
In children younger than 4 years, mortality related to the consequence of brainstem compression, which causes sudden death. In individuals aged 5-24 years, central nervous system (CNS) and respiratory abnormalities are the common causes of death. In patients aged 25-54 years, cardiovascular problems are the most frequent causes of death.
Morbidity associated with achondroplasia may include the following:
- Recurrent otitis media
- Hearing loss
- Neurologic complications due to cervicomedullary compression (eg, hypotonia, respiratory insufficiency, apnea, cyanotic episodes, feeding problems, quadriparesis)
- Obstructive and restrictive respiratory complications (eg, upper airway obstruction, pneumonia, apnea)
- Hydrocephalus
- Spinal deformities (eg, kyphosis, lordosis, scoliosis)
- Obesity
- Spinal canal stenosis
- Genu varum
- Cardiovascular complications
Children
- less muscle tone
- delayed walking skills
- delayed motor skills\
- bowed leg
- scoliosis
- lordosis
- arthritis
- issues with joint flexibility
- breathing problems
- ear infections
- crowded teeth
- Hydrocephalus
- obesity
- sleep apnea
- numbness in their legs
- tingling in their legs
- Women face higher risk pregnancies generally have their babies delivered through C-sections to prevent complications that could occur with a natural birth.
Prognosis
Overall prognosis of achondroplasia is good, with near-normal life expectancy in heterozygous individuals. When homozygous, the condition is fatal due to respiratory failure
Achondroplasia is a result of an autosomal dominant mutation in the fibroblast growth factor receptor gene 3 (FGFR3) located on chromosome 4p16.3 which causes an abnormality of cartilage formation. FGFR3 normally has a negative regulatory effect on bone growth. In achondroplasia, the mutated form of the receptor is constitutively active and this leads to severely shortened bones. All bones that form by endochondral ossification are affected. Bones that form by membranous ossification are not affected, thus allowing the skull vault to develop normally. People with achondroplasia have one normal copy of the fibroblast growth factor receptor 3 gene and one mutant copy. Two copies of the mutant gene are invariably fatal before, or shortly after birth. Only one copy of the gene needs to be present for the disorder to occur. Therefore, a person with achondroplasia has a 50% chance of passing on the gene to their offspring, meaning that there will be a 50% chance that each child will have achondroplasia. Since two copies (Homozygous) are fatal, if two people with achondroplasia have a child, there is a 25% chance of the child dying shortly after birth, a 50% chance the child will have achondroplasia, and a 25% chance the child will have a normal phenotype. However, in 3 out of 4 cases, people with achondroplasia are born to parents who don't have the condition. This is the result of a new mutation[33][14].
New gene mutations are associated with increasing paternal age (over 35 years). Studies have demonstrated that new gene mutations are exclusively inherited from the father and occur during spermatogenesis (as opposed to resulting from a gonadal mosaicism). More than 99% of achondroplasia is caused by two different mutations in the fibroblast growth factor receptor 3 (FGFR3). In about 98% of cases, a G to A point mutation at nucleotide 1138 of the FGFR3 gene causes a glycine to arginine substitution (Bellus et al 1995, Shiang et al 1994, Rousseau et al 1996). About 1% of cases are caused by a G to C point mutation at nucleotide 1138[14][24].
Biopsy from the growth plates of the ilium and proximal fibula revealed an essentially normal structure. Glycosaminoglycan determination is normal. The proportion of proteoglycan aggregates increases in the fibular head. The defect is mainly quantitative and lies in the proliferative zone of the growth plate.
There are two other syndromes with a genetic basis similar to achondroplasia: hypochondroplasia and thanatophoric dysplasia. Both of these disorders are also caused by a genetic mutation in the FGFR3 gene.
- SADDAN syndrome: severe achondroplasia with developmental delay and acanthosis nigricans
Symptoms
- Frequent ear infections (due to Eustachian tube blockages)
- Sleep apnea [27](which can be central or obstructive)
Physical Examination[33]
General
- Motor milestones may lag by 3-6 months
- Onset and frequent otitis media
- Conductive hearing loss
- Upper-airway obstruction, small chest wall, pectus excavatum
- Delayed gross motor development
- Achondroplastic dwarfs have short stature , with an average adult height of 131 cm (4 feet 3.8 inches) for males and 123 cm (4 feet 0.6 inches) for females.
- Stenosis of the spinal canal and intervertebral foramen
- low back pain, leg pain, dysesthesia, paresthesia, paraparesis, incontinence, and neurogenic claudication.
- Spinal kyphosis (convex curvature of the spine) or lordosis (concave curvature of the spine)
HEENT[24]
- Macrocephaly
- Hydrocephalus may be present
- A large head with prominent forehead
- Small midface or midface hypoplasia
- Flattened nasal bridge
Extremities
- Joint laxity
- Shortening of the proximal limbs (termed rhizomelic shortening)
- Short fingers and toes
- Varus (bowleg) or valgus (knock knee) deformities
- Fibromyalgia (trigger points located in the lower part of the back)
- trochanteric bursitis
Ultrasonography
The Ultrasonography may not be useful for diagnosing achondroplasia in the first half of the pregnancy; because in the fetus, heterozygous achondroplasia is associated with normal or near-normal femur lengths until 20-24 weeks of pregnancy. Later in the pregnancy, US evaluation can reveal short-limb dysplasia. The diagnosis can be made by fetal ultrasound by progressive discordance between the femur length and biparietal diameter by age. The trident hand configuration can be seen if the fingers are fully extended.
Radiography
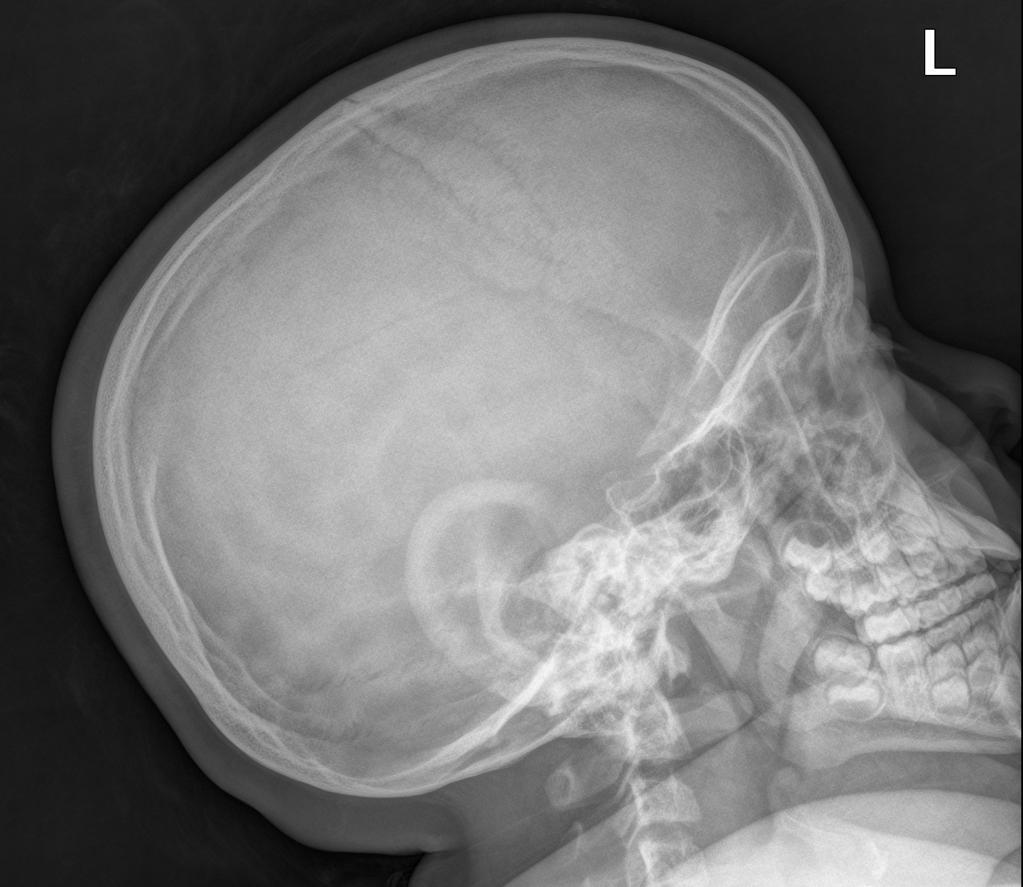
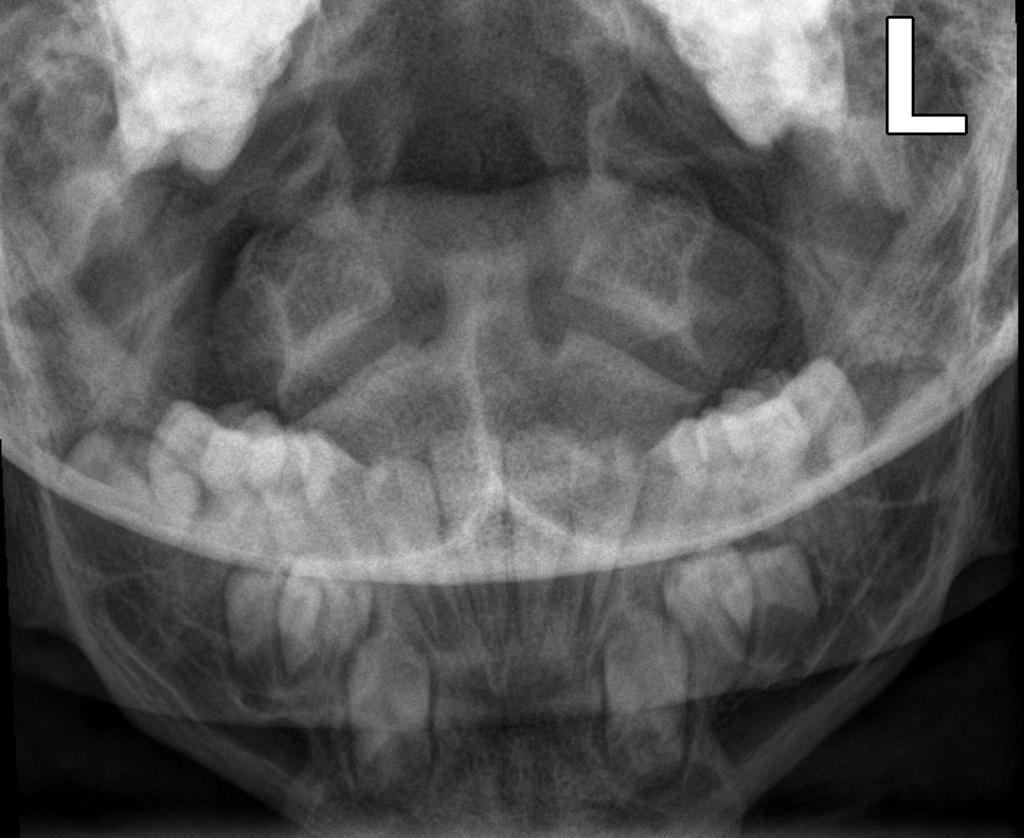
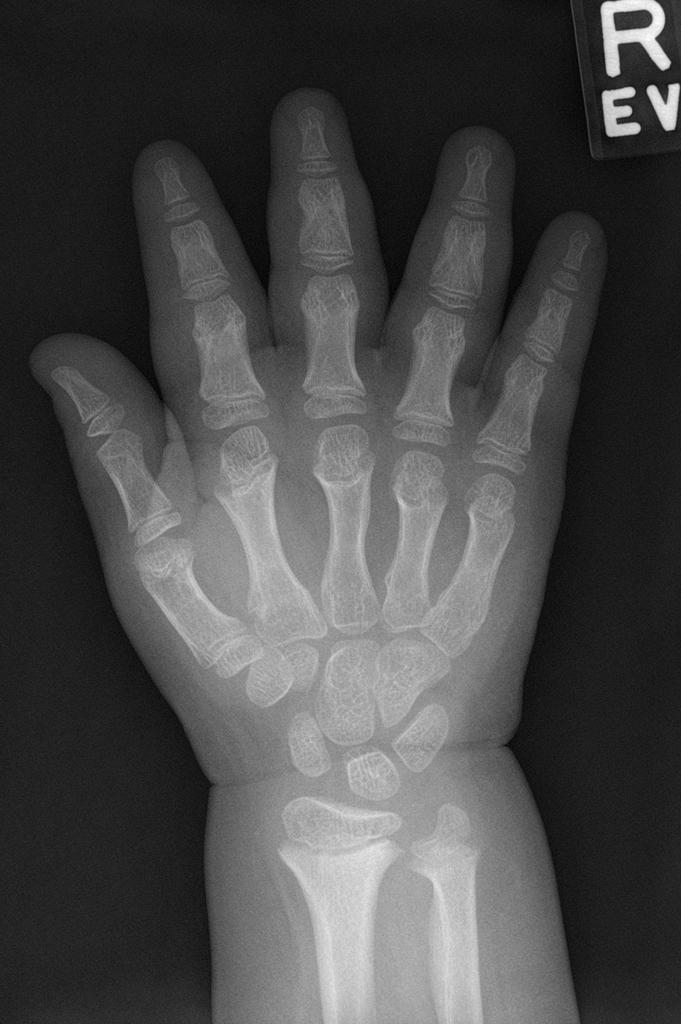
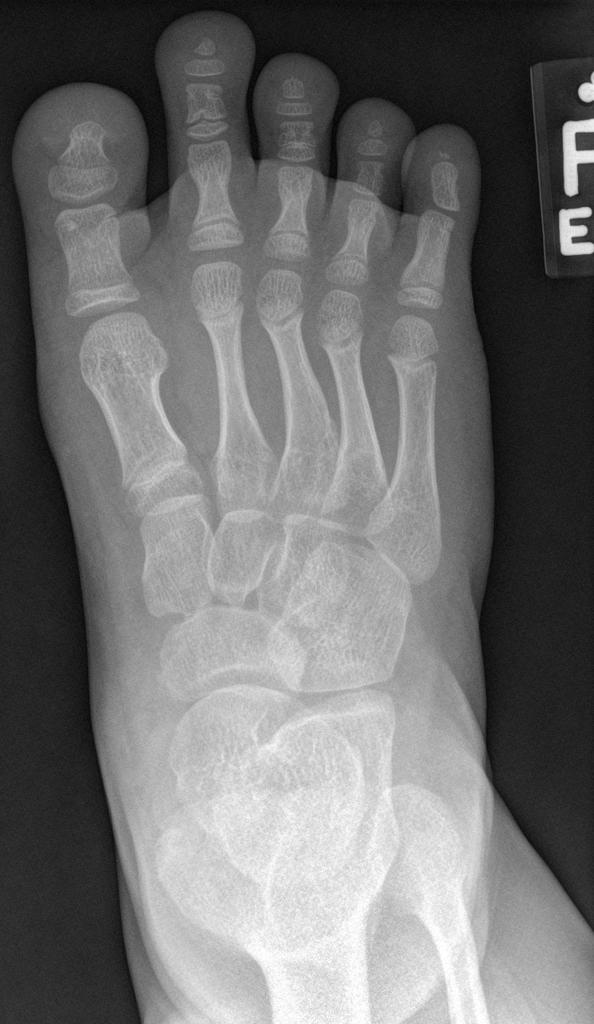
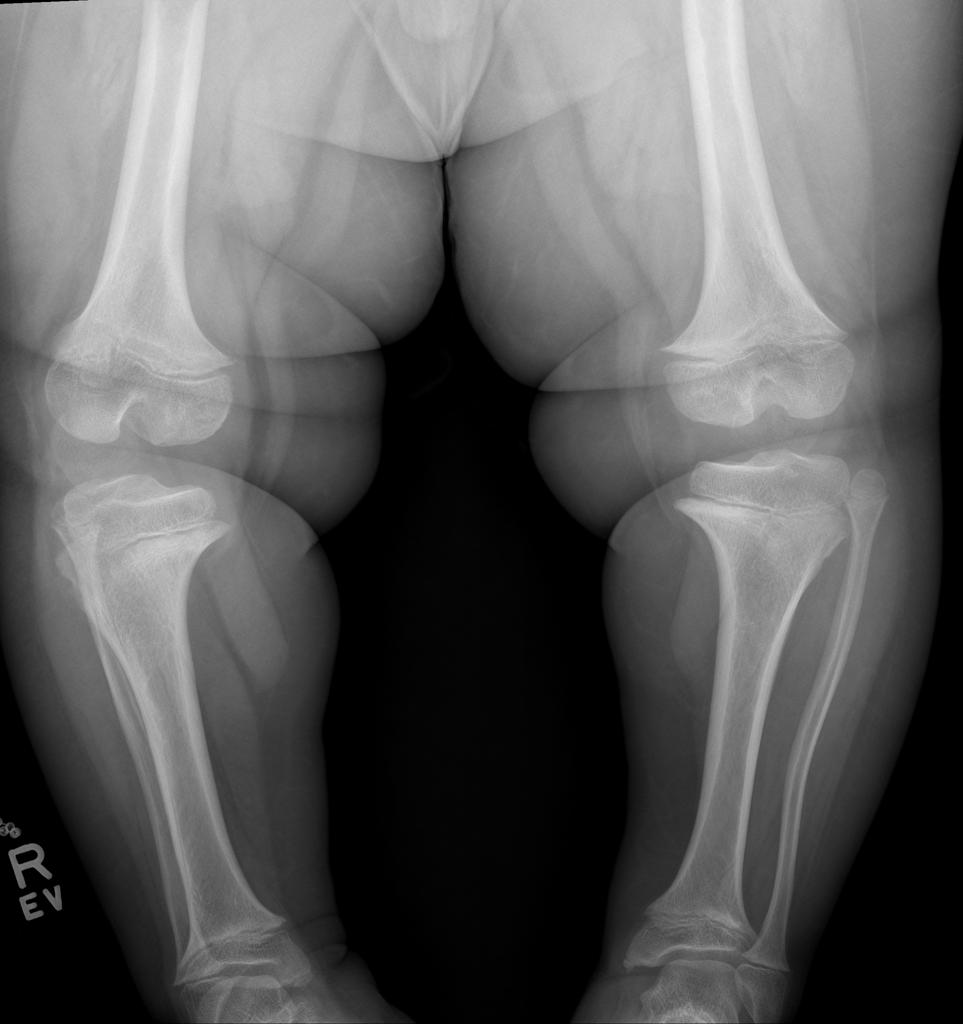
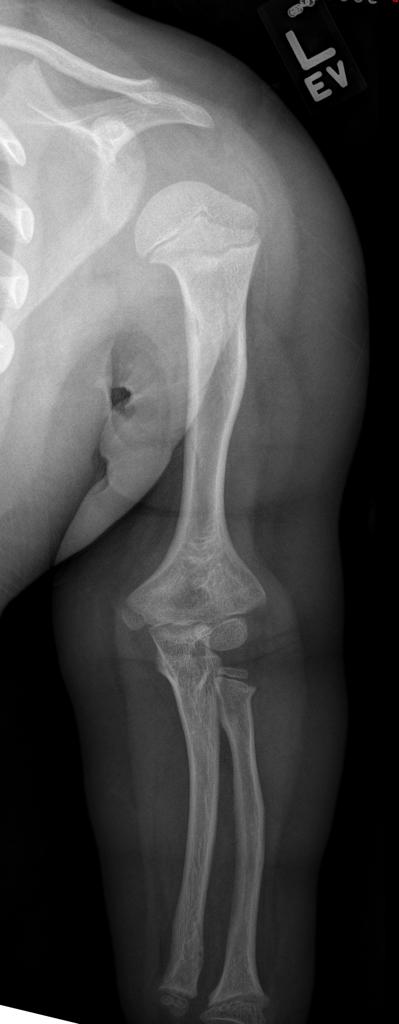
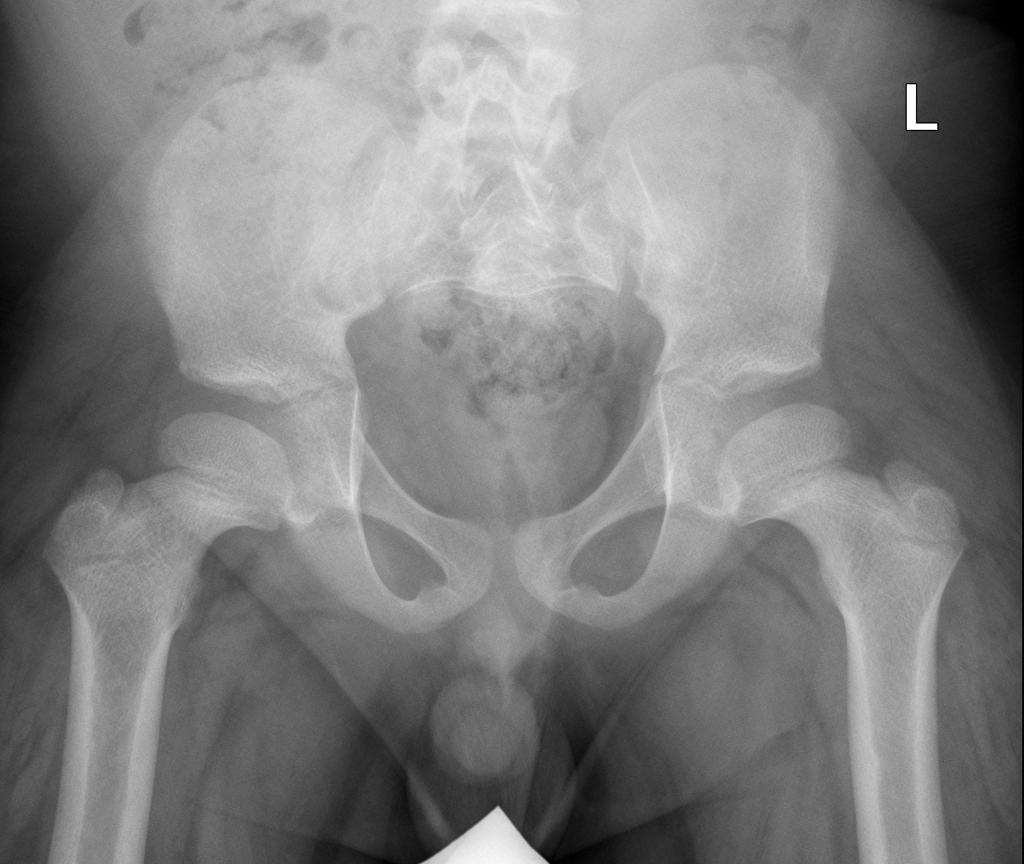
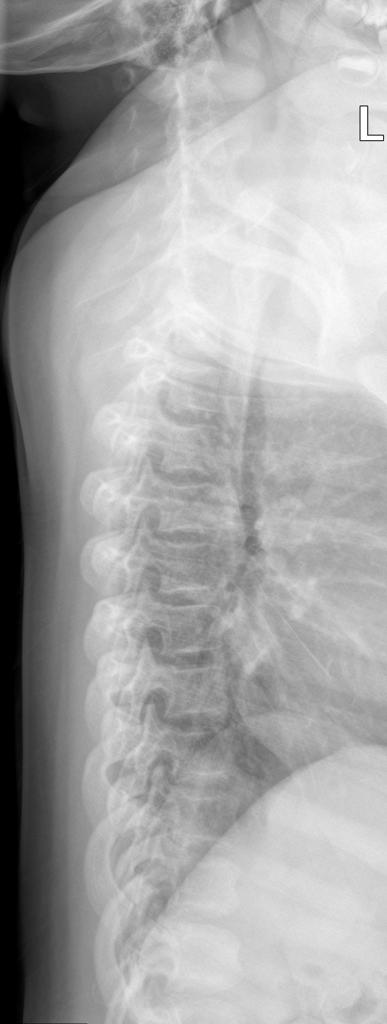
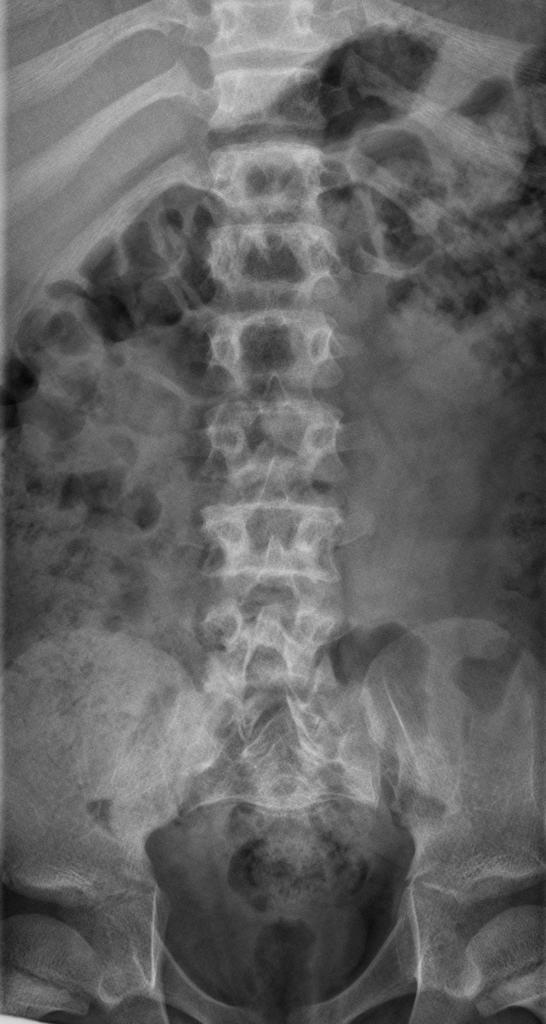
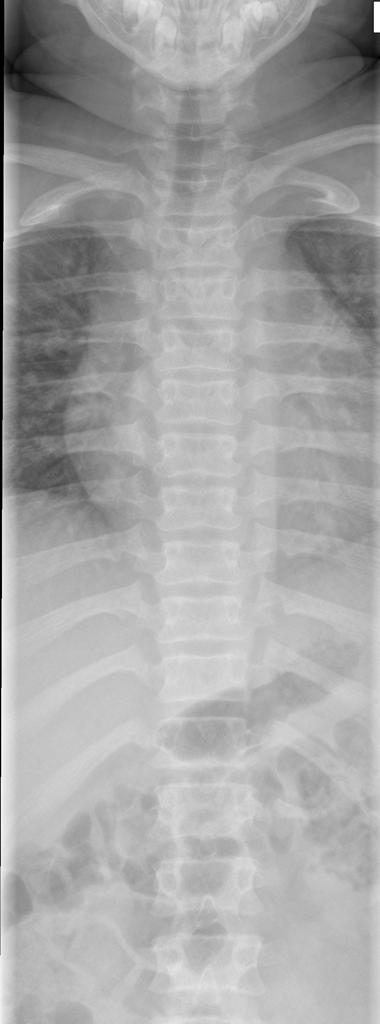
A skeletal survey is useful to confirm the diagnosis of achondroplasia. Radiographs of the skull, spine, and extremities show the specific features. A lateral skull radiograph demonstrates midface hypoplasia, enlarged calvaria, frontal prominence, and shortening of the base of the skull. Skull films demonstrate a large skull with a narrow foramen magnum, and relatively small skull base. The vertabral bodies are short and flattened with relatively large intervertebral disk height, and there is congenitally narrowed spinal canal. The iliac wingsare small and squared, with a narrow sciatic notch and horizontal acetabular roof. The tubular bones are short and thick with metaphyseal cupping and flaring and irregular growth plates. Fibular overgrowth is present. The hand is broad with short metacarpals and phalanges, and a trident configuration. The ribs are short with cupped anterior ends. If the radiographic features are not classic, a search for a different diagnosis should be entertained. Because of the extremely deformed bone structure, people with achondroplasia are often double jointed.
More obvious signs are a prominent forehead, flat nose bridge, protruding jaw, and crowded teeth.
- Shortening of tubular bones with a normal shaft caliber
- Short phalanges
- Squared iliac wings and narrow sacroiliac notch: champagne glass
- Fingers widely opposed and equal length: trident hands
- Enlarged skull vault and mandible
- Small foramen magnum
- Decreased lumber interpediculate distance and narrow spinal canal
Primary radiographic criteria for diagnosis include:
- Decrease in interpedicular distance in the lumbar spine
- Square short ilia
- Short, broad neck of femur
- Shortening of long tubular bones, with metaphyseal flaring
- Brachydactyly
Secondary radiographic criteria for diagnosis include:
- AP shortening of lumbar pedicles
- Dorsal concavity of lumbar vertebra
- Long distal fibula
- Short distal ulna
- Long ulnar styloid
Features on radiographs, CT, and MRI have similarities:
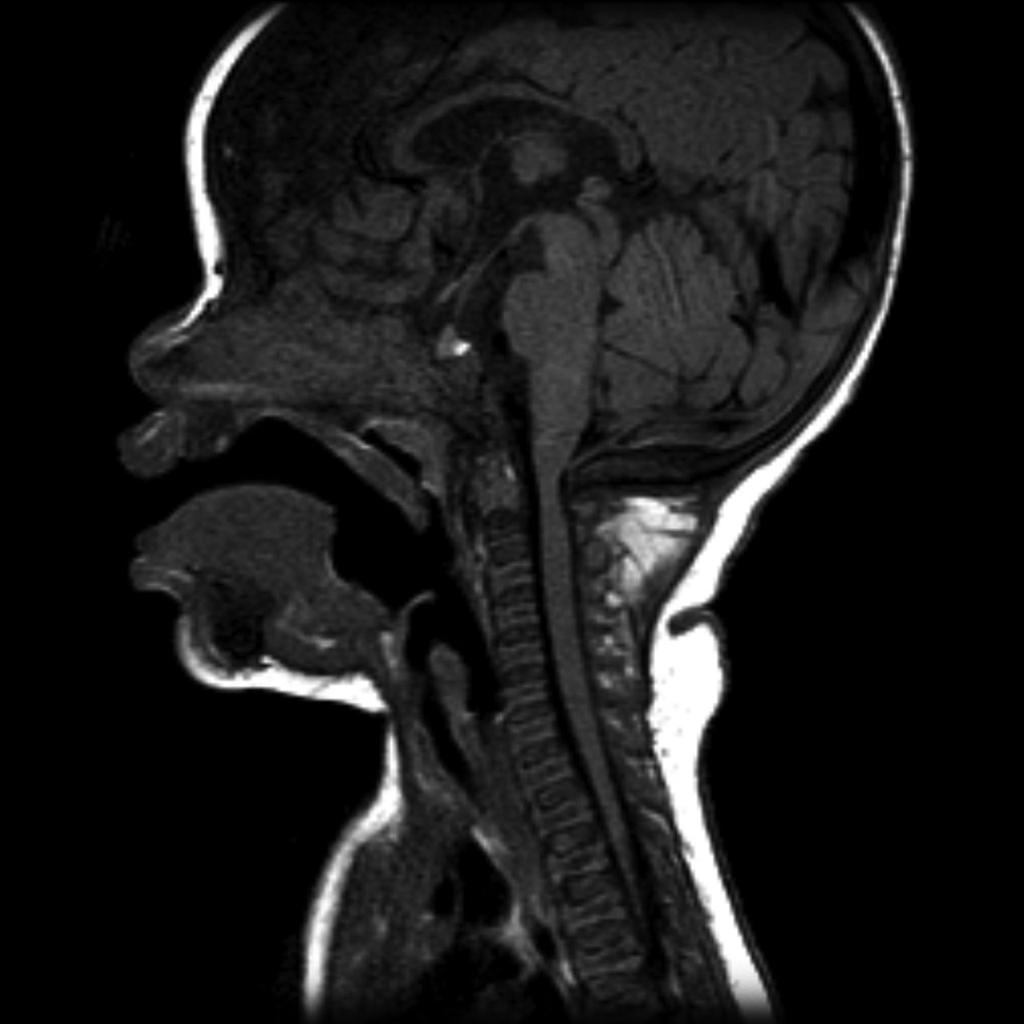
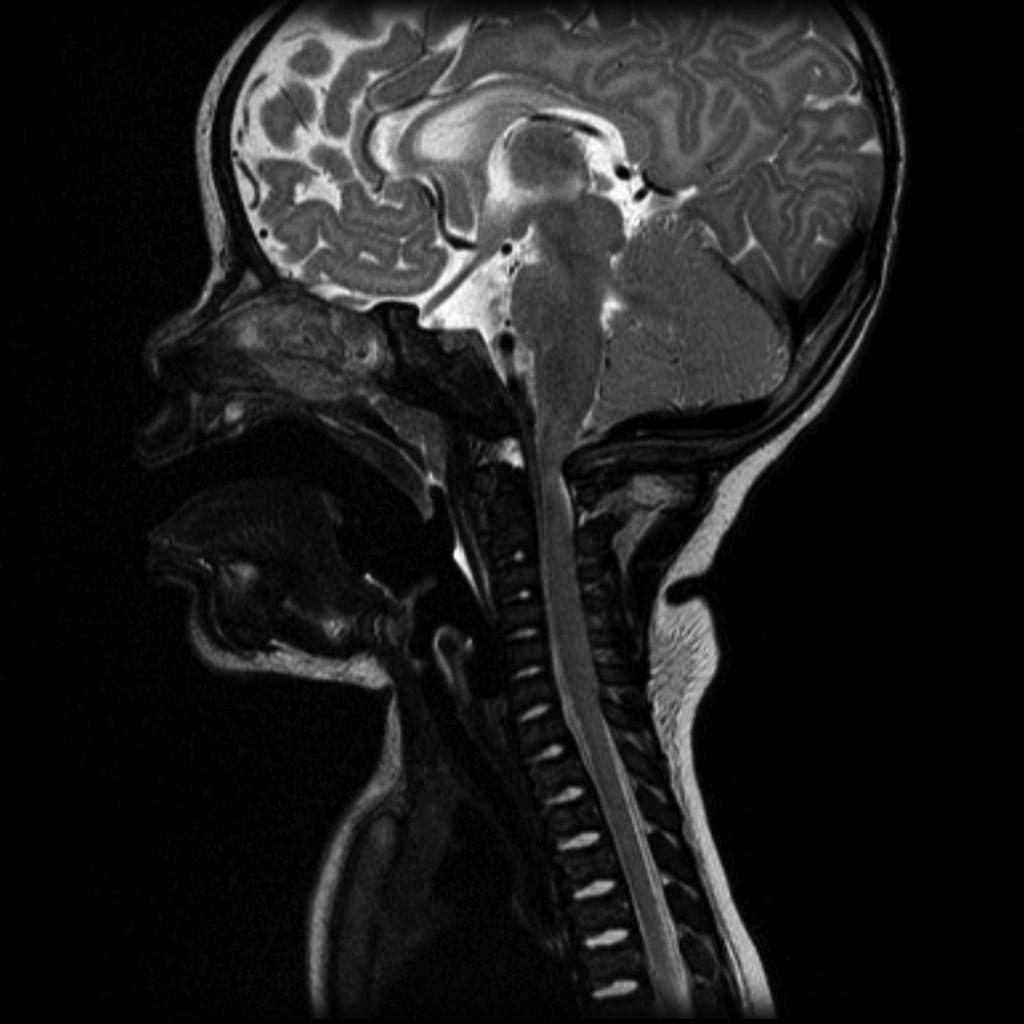
Cranial view
- relatively large cranial vault with small skull base
- prominent forehead with the depressed nasal bridge narrowed foramen magnum
- cervico-medullary kink
- relative elevation of the brainstem resulting in a large suprasellar cistern and vertically-oriented straight sinus [41]
- communicating hydrocephalus (caused by the venous obstruction at sigmoid sinus) [41]
Spinal view
- posterior vertebral scalloping
- progressive decrease in the interpedicular distance in the lumbar spine
- gibbus: thoracolumbar kyphosis with bullet-shaped/hypoplastic vertebra (not to be confused with Hurler syndrome)
- short pedicle canal stenosis
- laminar thickening
- widening of intervertebral diskss
- an increased angle between the sacrum and lumbar spine
Chest view
- anterior flaring of the ribs
- anteroposterior narrowing of the ribs
Pelvis and hips view
- horizontal acetabular roof (decreased acetabular angle)
- small squared (tombstone or mickey mouse ear) iliac wings
- small trident pelvis
- champagne glass type pelvic inlet
- short sacroiliac notches
Limbs view
- metaphyseal flaring: can give a trumpet bone type appearance
- the femora and humeri are particularly shortened (rhizomelic shortening)
- long fibula: the fibular head is at the level of the tibial plateau
- the limbs may also appear thickened but are in fact normal in absolute terms; thickening is perceived due to reduced length
- trident hand
- the metacarpal and metatarsal bones, and in some cases the proximal phalanges, are short and of similar length
Other
The Somatosensory evoked potential (SSEP) probably is related to brainstem compression at the level of the foramen magnum and its abnormalities reported around 44% of neurologically intact individuals with achondroplasia.
Since, the vital capacity is decreased, averaging 68% for affected males and 72% for affected females thepulmonary function tests are useful for preoperative evaluation when respiratory symptoms are present in these cases[26][42].
A sleep study may be performed if symptoms suggest airway obstruction.The cause of airway obstruction can be established to guide the treatment[38][24].
Treatment
At present, there is no actual treatment for achondroplasia. Related treatment varies and is usually managed by an orthopedic surgeon, particularly to correct kyphoscolioses, as well as neurosurgical, to decompress the foramen magnum or shunt hydrocephalus.In cases with achondroplasia the cervical cord compression due to narrowing of the foramen magnum is an important issue. Also, considering the sudden blindness associated with an increase in the intracranial pressure (ICP) the ICP monitoring ICP higher than 15 mm Hg is recommended in persons with achondroplasia with moderate ventriculomegaly, as demonstrated by MRI. This is performed with a percutaneous spinal catheter. Treatment is recommended when the ICP is higher than 15 mm Hg[43] [44][45][38]Closing </ref> missing for <ref> tag[46][36].
Medical Considerations
The somatotropin (recombinant human growth hormone [GH]) has revolutionized the treatment of short stature. GH is currently being used to augment the height of patients with achondroplasia. For maximum benefit, it is recommended that therapy be intiated at a young age (1-6 years)[43][45][47][42][48].
Surgical Considerations
Most of the orthopedic problems encountered in patients with achondroplasia are related to the spine[43][12][44]. Craniocervical stenosis, thoracolumbar kyphosis, spinal stenosis, angular deformities of the lower extremities, and lengthening of the short extremities are the orthopedic problems commonly addressed in achondroplasia [44][49][50][51].
- Treatment of spinal canal stenosis
- Treatment of thoracolumbar kyphosis
- Correction of genu varum
- Lengthening of limbs
- Foramen magnum decompression (neurosurgery)
Diet Considerations
Nutritional counseling is helpful. Obesity is a lifelong issue in this regard, and dietary therapy should be initiated early in life fro these cases[51].
.
-
 Case courtesy of Dr Hani Salam, <a href="https://radiopaedia.org/">Radiopaedia.org</a>. From the case <a href="https://radiopaedia.org/cases/9294">rID: 9294</a>
Case courtesy of Dr Hani Salam, <a href="https://radiopaedia.org/">Radiopaedia.org</a>. From the case <a href="https://radiopaedia.org/cases/9294">rID: 9294</a> -
-
-
-
-
-
-
-
-
-
-
-

.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
References
- ↑ Natt RS, Helliwell T, McCormick M (September 2009). "Tracheopathia chondro-osteoplastica--an unusual cause of stridor". J Laryngol Otol. 123 (9): 1039–41. doi:10.1017/S0022215108003721. PMID 18854058.
- ↑ Moreira Teixeira LS, Patterson J, Luyten FP (September 2014). "Skeletal tissue regeneration: where can hydrogels play a role?". Int Orthop. 38 (9): 1861–76. doi:10.1007/s00264-014-2402-2. PMID 24968789.
- ↑ Horton D, Anderson S, Hope NG (June 2014). "A review of current concepts in radiofrequency chondroplasty". ANZ J Surg. 84 (6): 412–6. doi:10.1111/ans.12130. PMID 23551491.
- ↑ Wu CC (January 2011). "Combined lateral retinacular release with drilling chondroplasty for treatment of patellofemoral osteoarthritis associated with patellar malalignment in elderly patients". Knee. 18 (1): 24–9. doi:10.1016/j.knee.2010.01.005. PMID 20171107.
- ↑ 5.0 5.1 Daugherty A (November 2017). "Achondroplasia: Etiology, Clinical Presentation, and Management". Neonatal Netw. 36 (6): 337–342. doi:10.1891/0730-0832.36.6.337. PMID 29185944.
- ↑ 6.0 6.1 6.2 Ornitz DM, Legeai-Mallet L (April 2017). "Achondroplasia: Development, pathogenesis, and therapy". Dev. Dyn. 246 (4): 291–309. doi:10.1002/dvdy.24479. PMC 5354942. PMID 27987249.
- ↑ 7.0 7.1 Unger S, Bonafé L, Gouze E (April 2017). "Current Care and Investigational Therapies in Achondroplasia". Curr Osteoporos Rep. 15 (2): 53–60. doi:10.1007/s11914-017-0347-2. PMC 5435778. PMID 28224446.
- ↑ 8.0 8.1 Shirley ED, Ain MC (April 2009). "Achondroplasia: manifestations and treatment". J Am Acad Orthop Surg. 17 (4): 231–41. PMID 19307672.
- ↑ 9.0 9.1 9.2 9.3 Dogba MJ, Rauch F, Douglas E, Bedos C (October 2014). "Impact of three genetic musculoskeletal diseases: a comparative synthesis of achondroplasia, Duchenne muscular dystrophy and osteogenesis imperfecta". Health Qual Life Outcomes. 12: 151. doi:10.1186/s12955-014-0151-y. PMC 4332447. PMID 25649344.
- ↑ 10.0 10.1 10.2 10.3 10.4 Tanaka H (October 2010). "[Cytokines in bone diseases. FGF receptor signaling and achondroplasia/hypochondroplasia]". Clin Calcium (in Japanese). 20 (10): 1490–6. doi:CliCa101014901496 Check
|doi=value (help). PMID 20890030. - ↑ Greening A, Mathews L, Blair J (December 2011). "Apparent dexmedetomidine-induced polyuric syndrome in an achondroplastic patient undergoing posterior spinal fusion". Anesth. Analg. 113 (6): 1381–3. doi:10.1213/ANE.0b013e31823299c1. PMID 22003216.
- ↑ 12.0 12.1 Bouali H, Latrech H (June 2015). "Achondroplasia: Current Options and Future Perspective". Pediatr Endocrinol Rev. 12 (4): 388–95. PMID 26182483.
- ↑ Mohindra S, Tripathi M, Arora S (2011). "Atlanto-axial instability in achondroplastic dwarfs: a report of two cases and literature review". Pediatr Neurosurg. 47 (4): 284–7. doi:10.1159/000335433. PMID 22472460.
- ↑ 14.0 14.1 14.2 Laederich MB, Horton WA (August 2010). "Achondroplasia: pathogenesis and implications for future treatment". Curr. Opin. Pediatr. 22 (4): 516–23. doi:10.1097/MOP.0b013e32833b7a69. PMID 20601886.
- ↑ 15.0 15.1 15.2 Accogli A, Pacetti M, Fiaschi P, Pavanello M, Piatelli G, Nuzzi D, Baldi M, Tassano E, Severino MS, Allegri A, Capra V (March 2015). "Association of achondroplasia with sagittal synostosis and scaphocephaly in two patients, an underestimated condition?". Am. J. Med. Genet. A. 167A (3): 646–52. doi:10.1002/ajmg.a.36933. PMID 25691418.
- ↑ 16.0 16.1 16.2 Zhao QH, Shi H, Hu JQ, Wang D, Fang G, Zhang YG, Wang YQ, Yang J (February 2017). "Ultrasound diagnosis of fetal thanatophoric skeletal dysplasia: Three cases report and a brief review". J. Huazhong Univ. Sci. Technol. Med. Sci. 37 (1): 148–152. doi:10.1007/s11596-017-1708-x. PMID 28224438.
- ↑ Yasoda A, Nakao K (August 2010). "[Genetic basis for skeletal disease. CNP therapy for achondroplasia]". Clin Calcium (in Japanese). 20 (8): 1212–8. doi:CliCa100812121218 Check
|doi=value (help). PMID 20675932. - ↑ de Azevedo Moreira LM, Matos MA, Schiper PP, Carvalho AF, Gomes IC, Rolemberg JC, Ferreira de Lima RL, Toralles MB (April 2010). "Co-occurrence of achondroplasia and Down syndrome: Genotype/phenotype association". Birth Defects Res. Part A Clin. Mol. Teratol. 88 (4): 228–31. doi:10.1002/bdra.20653. PMID 20222028.
- ↑ Ozono K, Namba N, Kubota T, Kitaoka T, Miura K, Ohata Y, Fujiwara M, Miyoshi Y, Michigami T (October 2012). "Pediatric aspects of skeletal dysplasia". Pediatr Endocrinol Rev. 10 Suppl 1: 35–43. PMID 23330244.
- ↑ 20.0 20.1 20.2 Pineau M, Farrow E, Nicot R, Ferri J (November 2018). "Achondroplasia: Orocraniofacial Features and Orthodontic-Surgical Management Guidelines Proposal". J Craniofac Surg. 29 (8): 2186–2191. doi:10.1097/SCS.0000000000004819. PMID 30277952.
- ↑ Stoilov I, Velinov M, Tsipuras P (1996). "[Achondroplasia--new research and future goals]". Mol Med (Sofia) (in Bulgarian). 1 (1): 13–7. PMID 8974759.
- ↑ Tanaka H (August 1997). "Achondroplasia: recent advances in diagnosis and treatment". Acta Paediatr Jpn. 39 (4): 514–20. PMID 9316303.
- ↑ Wilkie AO (April 2005). "Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations". Cytokine Growth Factor Rev. 16 (2): 187–203. doi:10.1016/j.cytogfr.2005.03.001. PMID 15863034.
- ↑ 24.0 24.1 24.2 24.3 Hasegawa K, Tanaka H (December 2014). "Children with short-limbed short stature in pediatric endocrinological services in Japan". Pediatr Int. 56 (6): 809–812. doi:10.1111/ped.12511. PMID 25244068.
- ↑ Vajo Z, Francomano CA, Wilkin DJ (February 2000). "The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans". Endocr. Rev. 21 (1): 23–39. doi:10.1210/edrv.21.1.0387. PMID 10696568.
- ↑ 26.0 26.1 26.2 Nikkel SM (October 2017). "Skeletal Dysplasias: What Every Bone Health Clinician Needs to Know". Curr Osteoporos Rep. 15 (5): 419–424. doi:10.1007/s11914-017-0392-x. PMID 28808977.
- ↑ 27.0 27.1 Zaffanello M, Cantalupo G, Piacentini G, Gasperi E, Nosetti L, Cavarzere P, Ramaroli DA, Mittal A, Antoniazzi F (February 2017). "Sleep disordered breathing in children with achondroplasia". World J Pediatr. 13 (1): 8–14. doi:10.1007/s12519-016-0051-9. PMID 27830579.
- ↑ Esteller E (2015). "[Obstructive sleep apnea-hypopnea syndrome in children: beyond adenotonsillar hypertrophy]". Acta Otorrinolaringol Esp (in Spanish; Castilian). 66 (2): 111–9. doi:10.1016/j.otorri.2014.05.001. PMID 25107357.
- ↑ Khetarpal P, Das S, Panigrahi I, Munshi A (February 2016). "Primordial dwarfism: overview of clinical and genetic aspects". Mol. Genet. Genomics. 291 (1): 1–15. doi:10.1007/s00438-015-1110-y. PMID 26323792.
- ↑ Alkuraya FS (February 2015). "Primordial dwarfism: an update". Curr Opin Endocrinol Diabetes Obes. 22 (1): 55–64. doi:10.1097/MED.0000000000000121. PMID 25490023.
- ↑ Yao G, Wang G, Wang D, Su G (January 2019). "Identification of a novel mutation of FGFR3 gene in a large Chinese pedigree with hypochondroplasia by next-generation sequencing: A case report and brief literature review". Medicine (Baltimore). 98 (4): e14157. doi:10.1097/MD.0000000000014157. PMC 6358355. PMID 30681580.
- ↑ Chen CP, Su YN, Lin TH, Chang TY, Su JW, Wang W (December 2013). "Detection of a de novo Y278C mutation in FGFR3 in a pregnancy with severe fetal hypochondroplasia: prenatal diagnosis and literature review". Taiwan J Obstet Gynecol. 52 (4): 580–5. doi:10.1016/j.tjog.2013.10.023. PMID 24411048.
- ↑ 33.0 33.1 Laederich MB, Horton WA (January 2012). "FGFR3 targeting strategies for achondroplasia". Expert Rev Mol Med. 14: e11. doi:10.1017/erm.2012.4. PMID 22559284.
- ↑ Blomberg M, Jeppesen EM, Skovby F, Benfeldt E (2010). "FGFR3 mutations and the skin: report of a patient with a FGFR3 gene mutation, acanthosis nigricans, hypochondroplasia and hyperinsulinemia and review of the literature". Dermatology (Basel). 220 (4): 297–305. doi:10.1159/000297575. PMID 20453470.
- ↑ Miura K, Oznono K (December 2013). "[Clinical condition and therapy of bone diseases]". Clin Calcium (in Japanese). 23 (12): 1789–94. doi:CliCa131217891794 Check
|doi=value (help). PMID 24292534. - ↑ 36.0 36.1 Martínez-Frías ML, de Frutos CA, Bermejo E, Nieto MA (January 2010). "Review of the recently defined molecular mechanisms underlying thanatophoric dysplasia and their potential therapeutic implications for achondroplasia". Am. J. Med. Genet. A. 152A (1): 245–55. doi:10.1002/ajmg.a.33188. PMID 20034074.
- ↑ Junxun L, Junru L, Meilan C, Chujia L, Shaoqian C, Jieyu Z, Zhuangjian Y, Fan Z, Juan O, Jing C, Juan L (November 2016). "Three patients with multiple myeloma developing secondary lymphoblastic leukemia: case reports and review of the literature". Tumori. 102 (Suppl. 2). doi:10.5301/tj.5000377. PMID 26166219.
- ↑ 38.0 38.1 38.2 Hecht JT, Bodensteiner JB, Butler IJ (2014). "Neurologic manifestations of achondroplasia". Handb Clin Neurol. 119: 551–63. doi:10.1016/B978-0-7020-4086-3.00036-9. PMID 24365319.
- ↑ McCarthy EF (February 2011). "Genetic diseases of bones and joints". Semin Diagn Pathol. 28 (1): 26–36. PMID 21675375.
- ↑ Cui Y, Zhao H, Liu Z, Liu C, Luan J, Zhou X, Han J (August 2012). "A systematic review of genetic skeletal disorders reported in Chinese biomedical journals between 1978 and 2012". Orphanet J Rare Dis. 7: 55. doi:10.1186/1750-1172-7-55. PMC 3492206. PMID 22913777.
- ↑ 41.0 41.1 Scott AA, Hodge KD, Torres-Martinez W, Dlouhy SR, Smith JL, Weaver DD (October 2017). "Sinus pericranii in achondroplasia: a case report and review of the literature". Clin. Dysmorphol. 26 (4): 252–255. doi:10.1097/MCD.0000000000000196. PMID 28872565.
- ↑ 42.0 42.1 Wit JM, Oostdijk W (June 2015). "Novel approaches to short stature therapy". Best Pract. Res. Clin. Endocrinol. Metab. 29 (3): 353–66. doi:10.1016/j.beem.2015.01.003. PMID 26051296.
- ↑ 43.0 43.1 43.2 Klag KA, Horton WA (April 2016). "Advances in treatment of achondroplasia and osteoarthritis". Hum. Mol. Genet. 25 (R1): R2–8. doi:10.1093/hmg/ddv419. PMID 26443596.
- ↑ 44.0 44.1 44.2 Wright MJ, Irving MD (February 2012). "Clinical management of achondroplasia". Arch. Dis. Child. 97 (2): 129–34. doi:10.1136/adc.2010.189092. PMID 21460402.
- ↑ 45.0 45.1 Afsharpaiman S, Saburi A, Waters KA (December 2013). "Respiratory difficulties and breathing disorders in achondroplasia". Paediatr Respir Rev. 14 (4): 250–5. doi:10.1016/j.prrv.2013.02.009. PMID 23523391.
- ↑ Schiedel F, Rödl R (2012). "Lower limb lengthening in patients with disproportionate short stature with achondroplasia: a systematic review of the last 20 years". Disabil Rehabil. 34 (12): 982–7. doi:10.3109/09638288.2011.631677. PMID 22112021.
- ↑ Miccoli M, Bertelloni S, Massart F (2016). "Height Outcome of Recombinant Human Growth Hormone Treatment in Achondroplasia Children: A Meta-Analysis". Horm Res Paediatr. 86 (1): 27–34. doi:10.1159/000446958. PMID 27355624.
- ↑ Sargar KM, Singh AK, Kao SC (October 2017). "Imaging of Skeletal Disorders Caused by Fibroblast Growth Factor Receptor Gene Mutations". Radiographics. 37 (6): 1813–1830. doi:10.1148/rg.2017170017. PMID 29019756.
- ↑ Paley D (May 2015). "PRECICE intramedullary limb lengthening system". Expert Rev Med Devices. 12 (3): 231–49. doi:10.1586/17434440.2015.1005604. PMID 25692375.
- ↑ Posey KL, Hecht JT (September 2017). "Novel therapeutic interventions for pseudoachondroplasia". Bone. 102: 60–68. doi:10.1016/j.bone.2017.03.045. PMC 6168010. PMID 28336490.
- ↑ 51.0 51.1 Krakow D (June 2015). "Skeletal dysplasias". Clin Perinatol. 42 (2): 301–19, viii. doi:10.1016/j.clp.2015.03.003. PMC 4456691. PMID 26042906.
de:Achondroplasie fa:آکندروپلازی gl:Acondroplasia ko:연골무형성증 nl:Achondroplasie sr:Ахондроплазија fi:Akondroplasia