Cystic fibrosis pathophysiology: Difference between revisions
(Created page with "{{CMG}} {{Cystic fibrosis}} ==Overview== ==Pathophysiology== Cystic fibrosis occurs when there is a mutation in the CFTR gene. The protein created by this gene is anchored t...") |
|||
| Line 5: | Line 5: | ||
==Pathophysiology== | ==Pathophysiology== | ||
Cystic fibrosis occurs when there is a mutation in the CFTR gene. The protein created by this gene is anchored to the [[cell membrane|outer membrane]] of [[cell (biology)|cell]]s in the [[sweat gland]]s, lungs, pancreas, and other affected [[organ (anatomy)|organ]]s. The protein spans this membrane and acts as a [[Ion channel|channel]] connecting the inner part of the cell ([[cytoplasm]]) to the [[extracellular fluid|surrounding fluid]]. In the airway this channel is primarily responsible for controlling the movement of chloride from inside to outside of the cell, however in the sweat ducts it facilitates the movement of chloride from the sweat into the cytoplasm. When the CFTR protein does not work, chloride is trapped inside the cells in the airway and outside in the skin. Because chloride is [[Electric charge|negatively charged]], positively charged [[ion]]s also cannot cross into the cell because they are affected by the [[Electrostatics|electrical attraction]] of the chloride ions. Sodium is the most common ion in the extracellular space and the combination of sodium and chloride creates the [[sodium chloride|salt]], which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test. | Cystic fibrosis occurs when there is a mutation in the CFTR gene. The protein created by this gene is anchored to the [[cell membrane|outer membrane]] of [[cell (biology)|cell]]s in the [[sweat gland]]s, lungs, pancreas, and other affected [[organ (anatomy)|organ]]s. The protein spans this membrane and acts as a [[Ion channel|channel]] connecting the inner part of the cell ([[cytoplasm]]) to the [[extracellular fluid|surrounding fluid]]. In the airway this channel is primarily responsible for controlling the movement of chloride from inside to outside of the cell, however in the sweat ducts it facilitates the movement of chloride from the sweat into the cytoplasm. When the CFTR protein does not work, chloride is trapped inside the cells in the airway and outside in the skin. Because chloride is [[Electric charge|negatively charged]], positively charged [[ion]]s also cannot cross into the cell because they are affected by the [[Electrostatics|electrical attraction]] of the chloride ions. Sodium is the most common ion in the extracellular space and the combination of sodium and chloride creates the [[sodium chloride|salt]], which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test. | ||
How this malfunction of cells in cystic fibrosis causes the clinical manifestations of CF is not well understood. One theory suggests that the lack of chloride exodus through the CFTR protein leads to the accumulation of more viscous, nutrient-rich mucus in the lungs that allows bacteria to hide from the body's [[immune system]]. Another theory proposes that the CFTR protein failure leads to a paradoxical increase in sodium and chloride uptake, which, by leading to increased water reabsorption, creates dehydrated and thick mucus. Yet another theory focuses on abnormal chloride movement ''out'' of the cell, which also leads to dehydration of mucus, pancreatic secretions, biliary secretions, etc. These theories all support the observation that the majority of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick faeces, etc.< | How this malfunction of cells in cystic fibrosis causes the clinical manifestations of CF is not well understood. One theory suggests that the lack of chloride exodus through the CFTR protein leads to the accumulation of more viscous, nutrient-rich mucus in the lungs that allows bacteria to hide from the body's [[immune system]]. Another theory proposes that the CFTR protein failure leads to a paradoxical increase in sodium and chloride uptake, which, by leading to increased water reabsorption, creates dehydrated and thick mucus. Yet another theory focuses on abnormal chloride movement ''out'' of the cell, which also leads to dehydration of mucus, pancreatic secretions, biliary secretions, etc. These theories all support the observation that the majority of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick faeces, etc. | ||
==Pathological Findings: A Case Example== | |||
===Clinical Summary=== | |||
A female infant was the product of an uncomplicated term delivery, though meconium staining was noted at birth. During the first day post-partum, the infant's abdomen became progressively distended and a meconium ileus was suspected. Surgery confirmed the presence of a meconium ileus and a section of perforated atretic jejunum proximal to the ileus was resected. Eight days later, the patient's condition had deteriorated. A second operation revealed a segment of necrotic bowel, which was removed. Subsequently the infant's pulmonary function deteriorated and she required frequent suctioning. She developed repeated episodes of pneumonia (E. coli and Pseudomonas grew out on cultures) complicated by atelectasis secondary to pneumothorax. The patient died at 25-days-of-age in respiratory failure. | |||
===Autopsy Findings=== | |||
Bilateral, extensive organizing bronchopneumonia was present with evidence of a pneumothorax and atelectasis. There were significant changes in the pancreas consistent with cystic fibrosis as well as involvement of the small intestine and changes related to the surgical procedures. | |||
[http://www.peir.net Images courtesy of Professor Peter Anderson DVM PhD and published with permission © PEIR, University of Alabama at Birmingham, Department of Pathology] | |||
<div align="left"> | |||
<gallery heights="175" widths="175"> | |||
Image:Cystic fibrosis 1.jpg|A gross photograph of liver and pancreas from the autopsy. The pancreas is slightly smaller than normal and it has a mucous consistency. | |||
Image:Cystic fibrosis 2.jpg|This section of duodenum demonstrates dilation, loss of rugae, and areas of ulceration (arrows). | |||
Image:Cystic fibrosis 3.jpg|This low-power photomicrograph of pancreas shows increased interstitial connective tissue resulting in accentuation of the lobular pattern. | |||
</gallery> | |||
</div> | |||
<div align="left"> | |||
<gallery heights="175" widths="175"> | |||
Image:Cystic fibrosis 4.jpg|This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected. | |||
Image:Cystic fibrosis 5.jpg|This higher-power photomicrograph shows a cystic space (1) within an acinar lobule. Islets of Langerhans (2) are also visible. | |||
Image:Cystic fibrosis 6.jpg|This high-power photomicrograph shows more clearly these variably-sized cystic spaces within the acinar pancreas. | |||
</gallery> | |||
</div> | |||
<div align="left"> | |||
<gallery heights="175" widths="175"> | |||
Image:Cystic fibrosis 7.jpg|This is another high-power photomicrograph showing cystic spaces (1) within the acinar pancreas and a normal islet of Langerhans (2). | |||
Image:Cystic fibrosis 8.jpg|This low-power photomicrograph of intestine shows the normal layers of the intestine, including the serosa (1), the muscularis (2), the submucosa (3), and the mucosal layer (4) with its deep mucosal crypts. There is yet another cystic space within the mucosa (5). | |||
Image:Cystic fibrosis 9.jpg|A higher-power photomicrograph shows the bottom of the intestinal crypts and the other normal layers of the intestine. Even at this magnification, accumulations of eosinophilic debris can be seen in many of the intestinal crypts (arrows). | |||
</gallery> | |||
</div> | |||
<div align="left"> | |||
<gallery heights="175" widths="175"> | |||
Image:Cystic fibrosis 10.jpg|This is a higher-power photomicrograph showing the eosinophilic debris in many of the intestinal crypts (arrows). | |||
Image:Cystic fibrosis 11.jpg|This higher-power photomicrograph shows more clearly the eosinophilic debris (arrows) in the intestinal crypts. | |||
Image:Cystic fibrosis 12.jpg|This is a low-power photomicrograph from another section of the intestine. Saggital sections of the intestinal crypts show the crypts along their full length, extending to the mucosal surface. | |||
</gallery> | |||
</div> | |||
<div align="left"> | |||
<gallery heights="175" widths="175"> | |||
Image:Cystic fibrosis 13.jpg|A higher-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows). | |||
Image:Cystic fibrosis 14.jpg|Another high-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows). | |||
</gallery> | |||
</div> | |||
==References== | ==References== | ||
{{Reflist|2}} | {{Reflist|2}} | ||
Revision as of 16:31, 3 February 2012
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
|
Cystic fibrosis Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Cystic fibrosis pathophysiology On the Web |
|
American Roentgen Ray Society Images of Cystic fibrosis pathophysiology |
|
Risk calculators and risk factors for Cystic fibrosis pathophysiology |
Overview
Pathophysiology
Cystic fibrosis occurs when there is a mutation in the CFTR gene. The protein created by this gene is anchored to the outer membrane of cells in the sweat glands, lungs, pancreas, and other affected organs. The protein spans this membrane and acts as a channel connecting the inner part of the cell (cytoplasm) to the surrounding fluid. In the airway this channel is primarily responsible for controlling the movement of chloride from inside to outside of the cell, however in the sweat ducts it facilitates the movement of chloride from the sweat into the cytoplasm. When the CFTR protein does not work, chloride is trapped inside the cells in the airway and outside in the skin. Because chloride is negatively charged, positively charged ions also cannot cross into the cell because they are affected by the electrical attraction of the chloride ions. Sodium is the most common ion in the extracellular space and the combination of sodium and chloride creates the salt, which is lost in high amounts in the sweat of individuals with CF. This lost salt forms the basis for the sweat test.
How this malfunction of cells in cystic fibrosis causes the clinical manifestations of CF is not well understood. One theory suggests that the lack of chloride exodus through the CFTR protein leads to the accumulation of more viscous, nutrient-rich mucus in the lungs that allows bacteria to hide from the body's immune system. Another theory proposes that the CFTR protein failure leads to a paradoxical increase in sodium and chloride uptake, which, by leading to increased water reabsorption, creates dehydrated and thick mucus. Yet another theory focuses on abnormal chloride movement out of the cell, which also leads to dehydration of mucus, pancreatic secretions, biliary secretions, etc. These theories all support the observation that the majority of the damage in CF is due to blockage of the narrow passages of affected organs with thickened secretions. These blockages lead to remodeling and infection in the lung, damage by accumulated digestive enzymes in the pancreas, blockage of the intestines by thick faeces, etc.
Pathological Findings: A Case Example
Clinical Summary
A female infant was the product of an uncomplicated term delivery, though meconium staining was noted at birth. During the first day post-partum, the infant's abdomen became progressively distended and a meconium ileus was suspected. Surgery confirmed the presence of a meconium ileus and a section of perforated atretic jejunum proximal to the ileus was resected. Eight days later, the patient's condition had deteriorated. A second operation revealed a segment of necrotic bowel, which was removed. Subsequently the infant's pulmonary function deteriorated and she required frequent suctioning. She developed repeated episodes of pneumonia (E. coli and Pseudomonas grew out on cultures) complicated by atelectasis secondary to pneumothorax. The patient died at 25-days-of-age in respiratory failure.
Autopsy Findings
Bilateral, extensive organizing bronchopneumonia was present with evidence of a pneumothorax and atelectasis. There were significant changes in the pancreas consistent with cystic fibrosis as well as involvement of the small intestine and changes related to the surgical procedures.
-
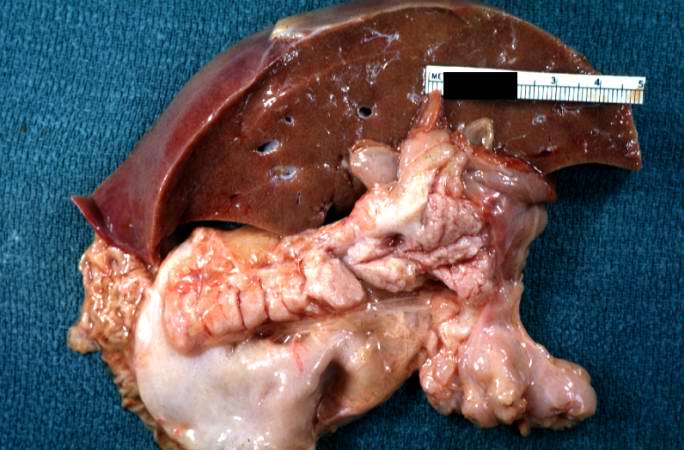
A gross photograph of liver and pancreas from the autopsy. The pancreas is slightly smaller than normal and it has a mucous consistency.
-
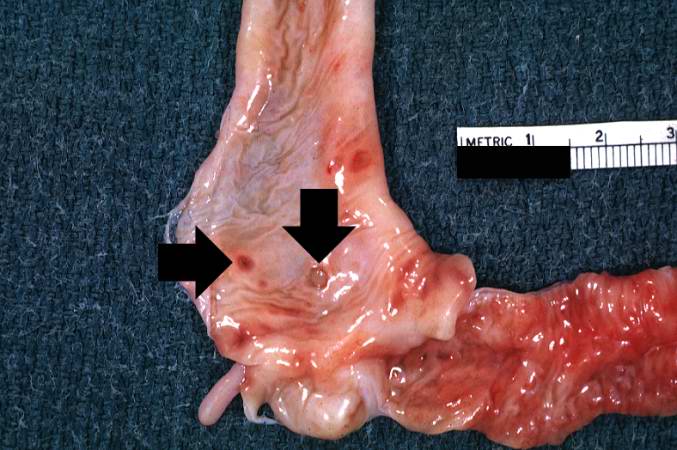
This section of duodenum demonstrates dilation, loss of rugae, and areas of ulceration (arrows).
-
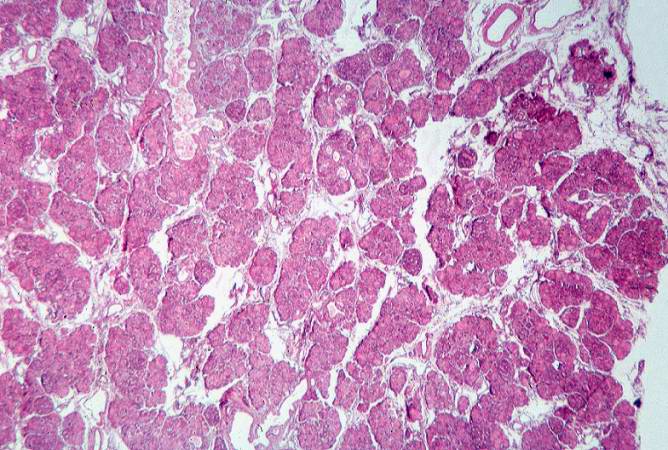
This low-power photomicrograph of pancreas shows increased interstitial connective tissue resulting in accentuation of the lobular pattern.



-
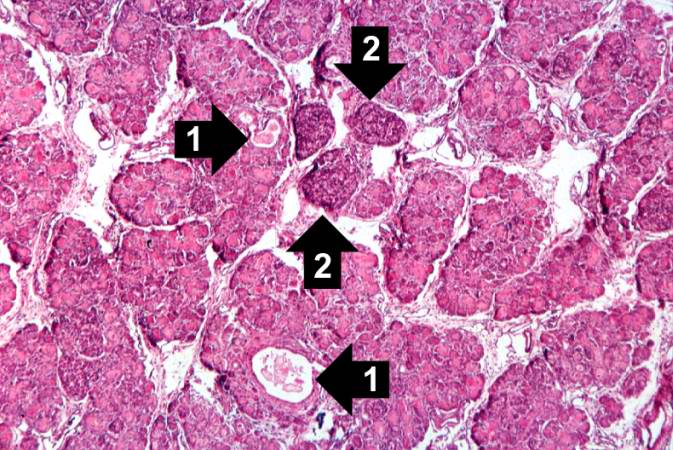
This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected.
-
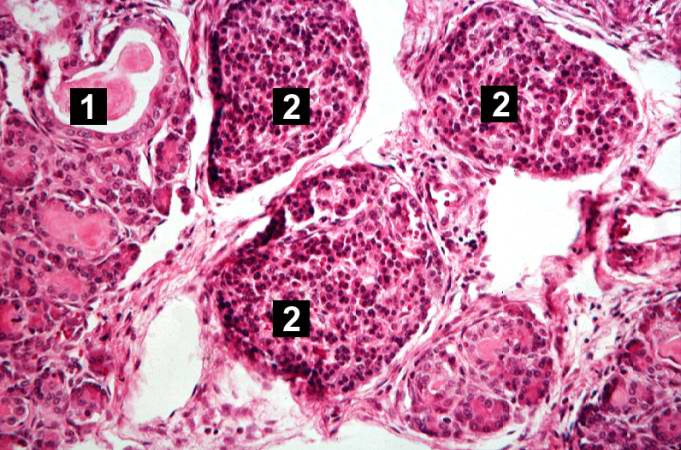
This higher-power photomicrograph shows a cystic space (1) within an acinar lobule. Islets of Langerhans (2) are also visible.
-
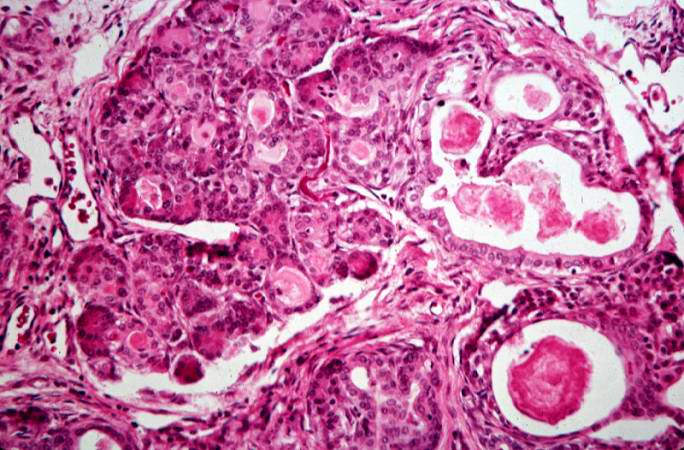
This high-power photomicrograph shows more clearly these variably-sized cystic spaces within the acinar pancreas.



-
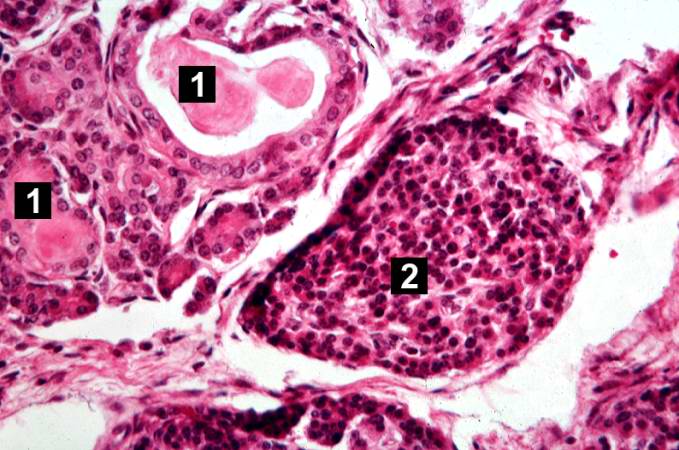
This is another high-power photomicrograph showing cystic spaces (1) within the acinar pancreas and a normal islet of Langerhans (2).
-
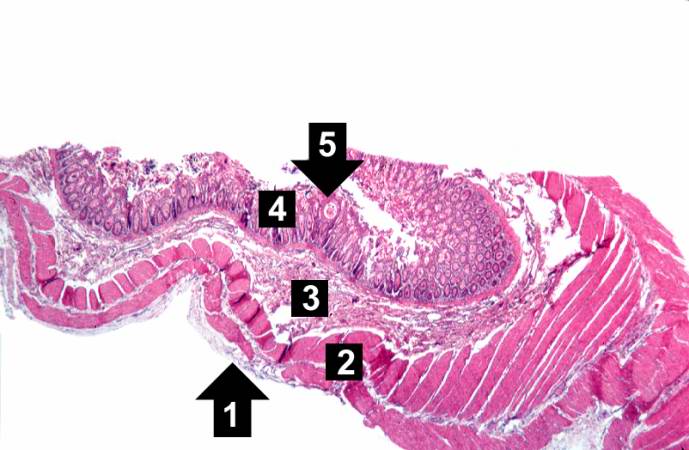
This low-power photomicrograph of intestine shows the normal layers of the intestine, including the serosa (1), the muscularis (2), the submucosa (3), and the mucosal layer (4) with its deep mucosal crypts. There is yet another cystic space within the mucosa (5).
-
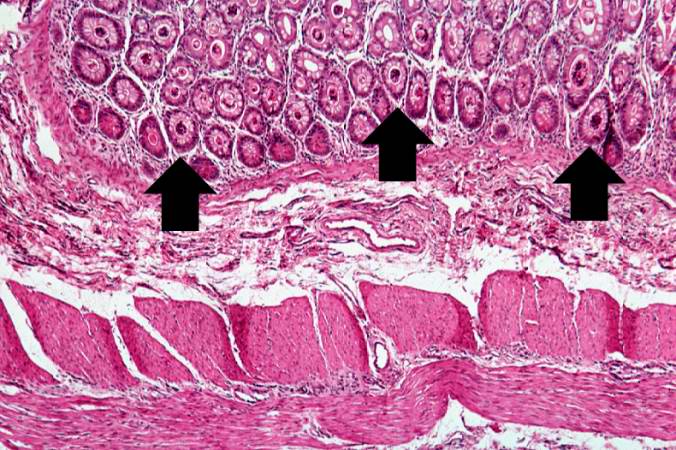
A higher-power photomicrograph shows the bottom of the intestinal crypts and the other normal layers of the intestine. Even at this magnification, accumulations of eosinophilic debris can be seen in many of the intestinal crypts (arrows).



-
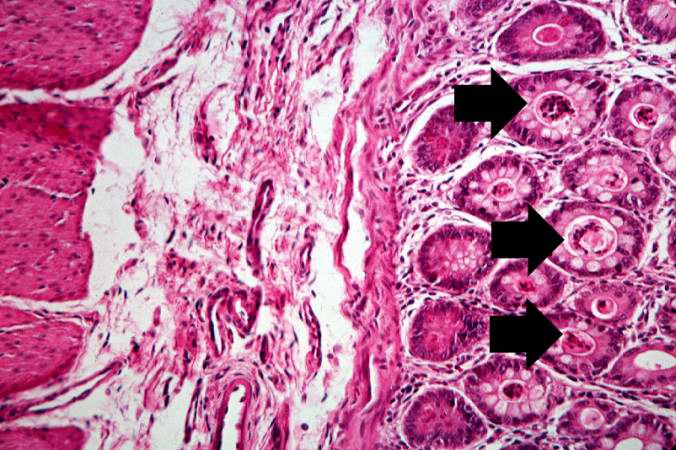
This is a higher-power photomicrograph showing the eosinophilic debris in many of the intestinal crypts (arrows).
-
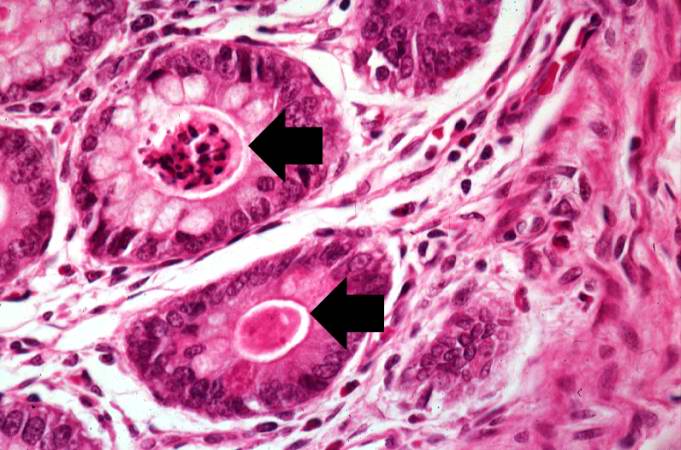
This higher-power photomicrograph shows more clearly the eosinophilic debris (arrows) in the intestinal crypts.
-
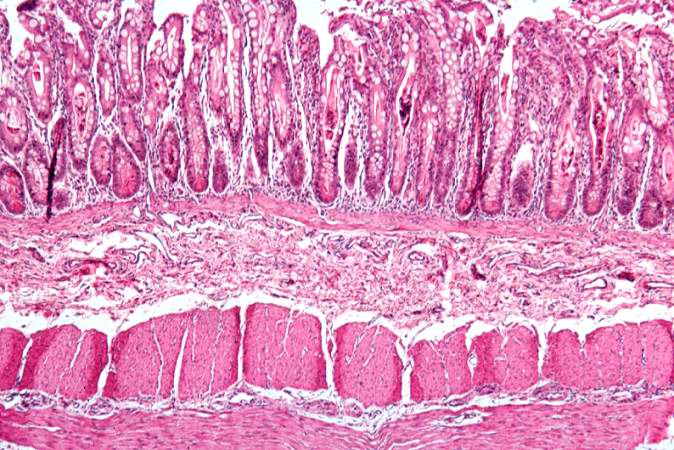
This is a low-power photomicrograph from another section of the intestine. Saggital sections of the intestinal crypts show the crypts along their full length, extending to the mucosal surface.



-
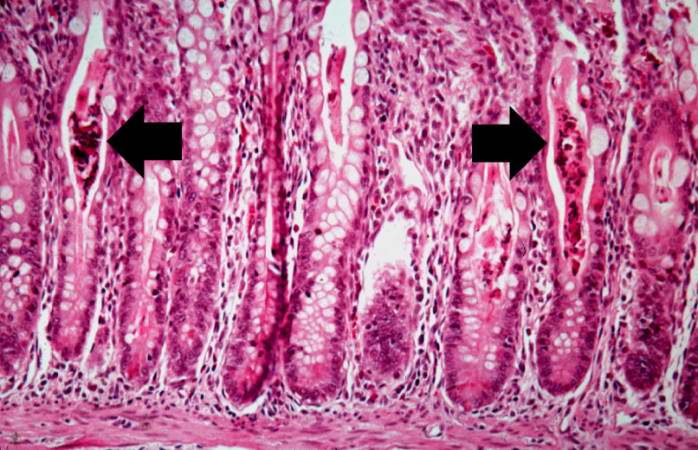
A higher-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).
-
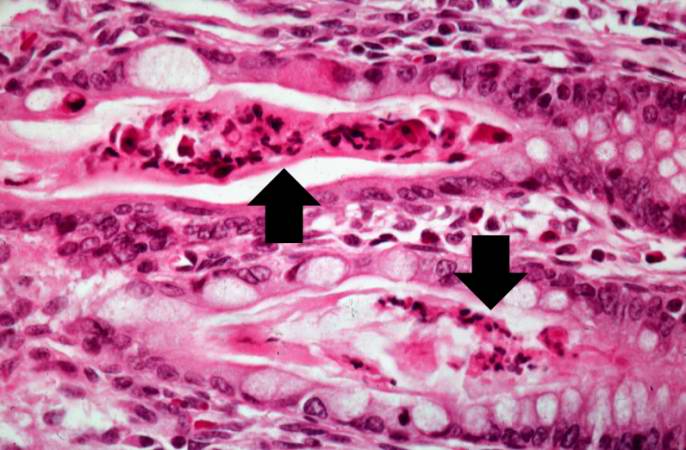
Another high-power photomicrograph of intestine shows the vacuolated intestinal epithelial cells lining the crypts and necrotic debris and inspissated secretions within the crypts (arrows).

