Medullary thyroid cancer pathophysiology: Difference between revisions
| Line 11: | Line 11: | ||
==Genetics== | ==Genetics== | ||
* Mutations in the [[RET proto-oncogene|''RET'']] (REarranged during Transfection), located on chromosome 10, lead to the [[gene expression|expression]] of a mutated [[receptor tyrosine kinase]] protein. [[RET proto-oncogene|''RET'']] is involved in the regulation of cell growth and development and its germline mutation is responsible for nearly all cases of hereditary or familial medullary thyroid carcinoma. Germline mutation of [[RET proto-oncogene|''RET'']] may also be responsible for the development of [[hyperparathyroidism]] and [[pheochromocytoma]]. Hereditary medullary thyroid cancer is inherited as an autosomal dominant trait, meaning that each child of an affected parent has a 50% probability of inheriting the mutant [[RET proto-oncogene|''RET'']] from the affected parent. DNA analysis makes it possible to identify children who carry the mutant gene. Hereditary medullary thyroid carcinoma or multiple endocrine neoplasia ([[Multiple endocrine neoplasia type 2|MEN2]]) accounts for approximately 25% of all medullary thyroid carcinomas. | * Mutations in the [[RET proto-oncogene|''RET'' proto-oncogene]] (REarranged during Transfection), located on chromosome 10, lead to the [[gene expression|expression]] of a mutated [[receptor tyrosine kinase]] protein. [[RET proto-oncogene|''RET'' proto-oncogene]] is involved in the regulation of cell growth and development and its germline mutation is responsible for nearly all cases of hereditary or familial medullary thyroid carcinoma. Germline mutation of [[RET proto-oncogene|''RET'']] may also be responsible for the development of [[hyperparathyroidism]] and [[pheochromocytoma]]. Hereditary medullary thyroid cancer is inherited as an autosomal dominant trait, meaning that each child of an affected parent has a 50% probability of inheriting the mutant [[RET proto-oncogene|''RET'']] from the affected parent. DNA analysis makes it possible to identify children who carry the mutant gene. Hereditary medullary thyroid carcinoma or multiple endocrine neoplasia ([[Multiple endocrine neoplasia type 2|MEN2]]) accounts for approximately 25% of all medullary thyroid carcinomas. | ||
* Seventy-five percent of medullary thyroid carcinoma occurs in individuals without an identifiable family history and is assigned the term "sporadic". Individuals who develop sporadic medullary thyroid carcinoma tend to be older and have more extensive disease at the time of initial presentation than those with a family history (screening is likely to be initiated at an early age in the hereditary form). Approximately 25-60% of sporadic medullary thyroid carcinomas have a somatic mutation (one that occurs within a single "parafollicular" cell) of the [[RET proto-oncogene|''RET'']]. | * Seventy-five percent of medullary thyroid carcinoma occurs in individuals without an identifiable family history and is assigned the term "sporadic". Individuals who develop sporadic medullary thyroid carcinoma tend to be older and have more extensive disease at the time of initial presentation than those with a family history (screening is likely to be initiated at an early age in the hereditary form). Approximately 25-60% of sporadic medullary thyroid carcinomas have a somatic mutation (one that occurs within a single "parafollicular" cell) of the [[RET proto-oncogene|''RET'']]. | ||
Revision as of 21:47, 14 December 2015
|
Medullary thyroid cancer Microchapters |
|
Differentiating Medullary thyroid cancer from other Diseases |
|---|
|
Diagnosis |
|
Treatment |
|
Case Studies |
|
Medullary thyroid cancer pathophysiology On the Web |
|
American Roentgen Ray Society Images of Medullary thyroid cancer pathophysiology |
|
Risk calculators and risk factors for Medullary thyroid cancer pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Ammu Susheela, M.D. [2]
Overview
Development of medullary thyroid cancer is the result of genetic mutation of RET proto-oncogene. On gross pathology, well circumscribed, gray, white, or yellow colored masses are characteristic findings of medullary thyroid cancer. On microscopic histopathological analysis, polygonal to spindle to small cells, interstitial edema, and vascular hyalinized stroma are characteristic findings of medullary thyroid cancer.
Pathogenesis
- Medullary tumors are the third most common of all thyroid cancers.
- Medullary thyroid carcinoma is a subtype of thyroid cancer which accounts for 5-10% of all thyroid malignancies.
- Medullary thyroid cancer is a form of thyroid carcinoma which originates from the parafollicular cells (C cells), which produce the hormone calcitonin.[1]It is characterized by consistent production of a hormonal marker called calcitonin. Medullary thyroid carcinoma is also characterized by calcification of both primary and metastatic sites. Metastatic involvement may be seen in up to 50% at the time of presentation.
- Approximately 25% of reported cases of medullary thyroid carcinoma are familial. Familial medullary thyroid carcinoma syndromes include multiple endocrine neoplasia type 2A, multiple endocrine neoplasia type 2B, and familial non-multiple endocrine neoplasia syndromes. Medullary thyroid carcinoma can secrete calcitonin and other peptide substances. Familial medullary thyroid carcinoma which constitute approximately 25% of medullary thyroid cancer is genetic in nature, caused by a mutation in the RET proto-oncogene. When medullary thyroid carcinoma occurs by itself it is termed sporadic medullary thyroid carcinoma.
Genetics
- Mutations in the RET proto-oncogene (REarranged during Transfection), located on chromosome 10, lead to the expression of a mutated receptor tyrosine kinase protein. RET proto-oncogene is involved in the regulation of cell growth and development and its germline mutation is responsible for nearly all cases of hereditary or familial medullary thyroid carcinoma. Germline mutation of RET may also be responsible for the development of hyperparathyroidism and pheochromocytoma. Hereditary medullary thyroid cancer is inherited as an autosomal dominant trait, meaning that each child of an affected parent has a 50% probability of inheriting the mutant RET from the affected parent. DNA analysis makes it possible to identify children who carry the mutant gene. Hereditary medullary thyroid carcinoma or multiple endocrine neoplasia (MEN2) accounts for approximately 25% of all medullary thyroid carcinomas.
- Seventy-five percent of medullary thyroid carcinoma occurs in individuals without an identifiable family history and is assigned the term "sporadic". Individuals who develop sporadic medullary thyroid carcinoma tend to be older and have more extensive disease at the time of initial presentation than those with a family history (screening is likely to be initiated at an early age in the hereditary form). Approximately 25-60% of sporadic medullary thyroid carcinomas have a somatic mutation (one that occurs within a single "parafollicular" cell) of the RET.
Associated Conditions
Gross Pathology
- Medullary thyroid cancer is usually a well circumscribed, gray, white, or yellow colored mass which is gritty to firm in consistency.
Microscopic Pathology
- Microscopic features of medullary thyroid cancer is as follows:
Cytoplasm and Nuclei
- Nested with delicate vascular septa
- Trabecular cells
- Tubular/glandular, pseudo-papillary cells
- Polygonal to spindle to small cells
- Amphophilic, somewhat granular cytoplasm
- Interstitial edema
- Stroma may have amyloid deposits with fluffy appearing acellular eosinophilic material in the cytoplasm
- Stroma is vascular and can show hemorrhage, hyalinised collagen, oedema or metaplastic bone
- Coarse calcification and psammoma bodies may be present
- Nuclei with neuroendocrine features
- Small, round nuclei.
- Coarse chromatin (salt and pepper nuclei)
Surrounding Thyroid
- +/- C-cell hyperplasia - seen with familial forms of medullary thyroid carcinoma
- C cells (AKA parafollicular cell): abundant cytoplasm - clear/pale
-
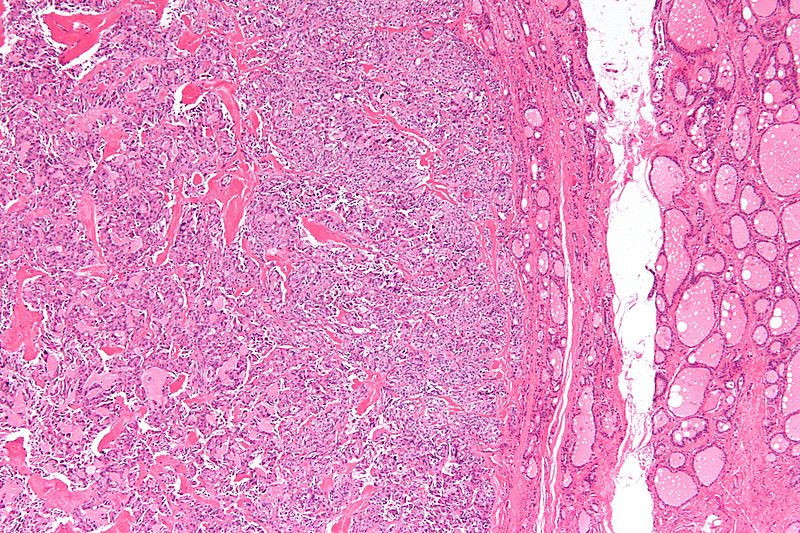
Low magnification micrograph of medullary thyroid carcinoma<ref> Medullary thyroid cancer librepathology
-
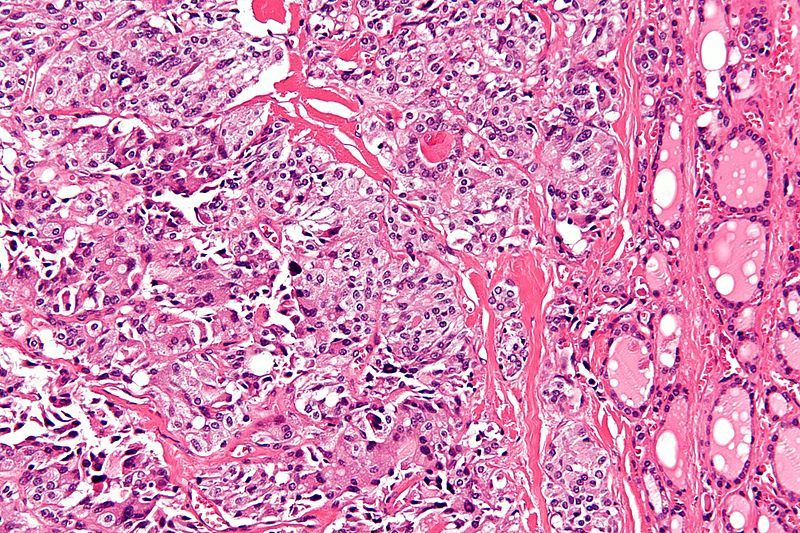
High magnification micrograph of medullary thyroid carcinoma<ref> Medullary thyroid cancer librepathology
-
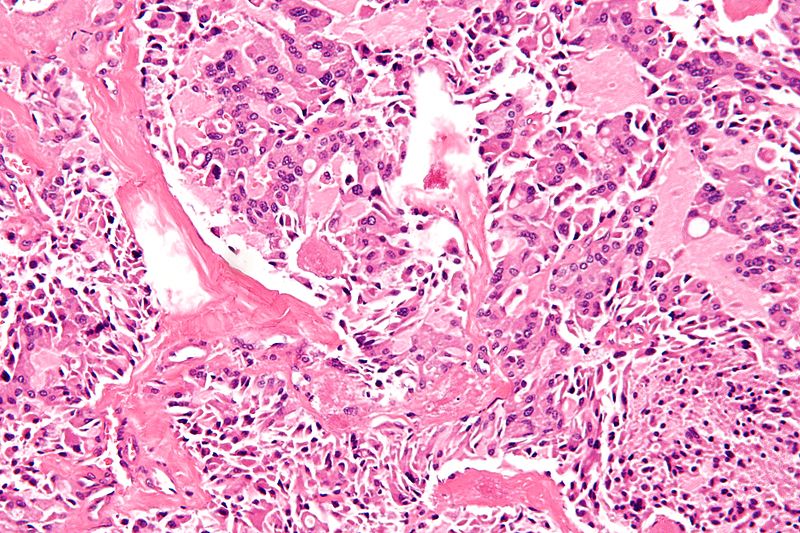
High magnification micrograph of medullary thyroid carcinoma<ref> Medullary thyroid cancer librepathology
-
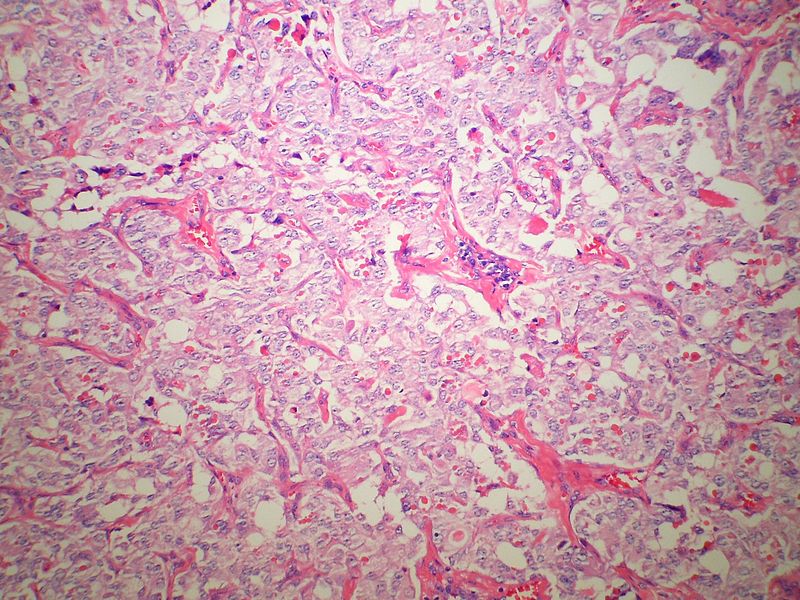
Low magnification micrograph of medullary thyroid carcinoma<ref> Medullary thyroid cancer librepathology
-
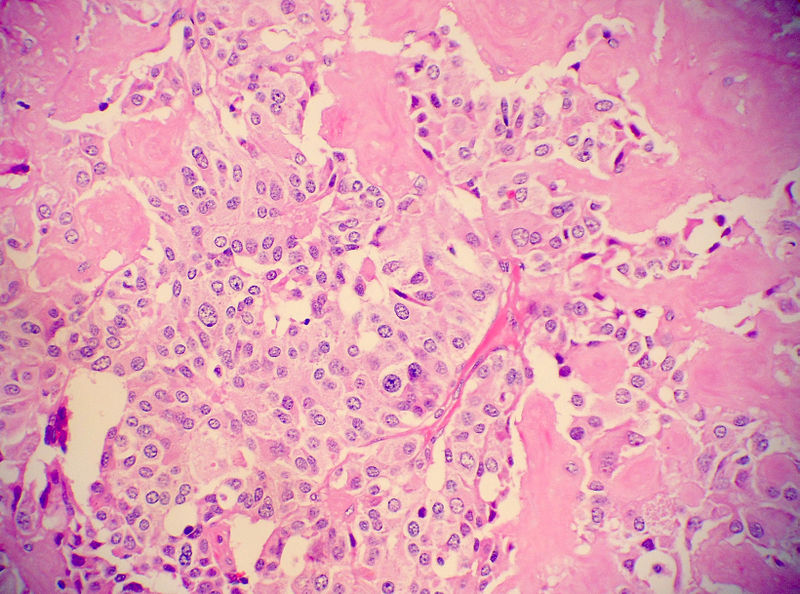
Medullary thyroid carcinoma amyloid<ref> Medullary thyroid cancer librepathology
-
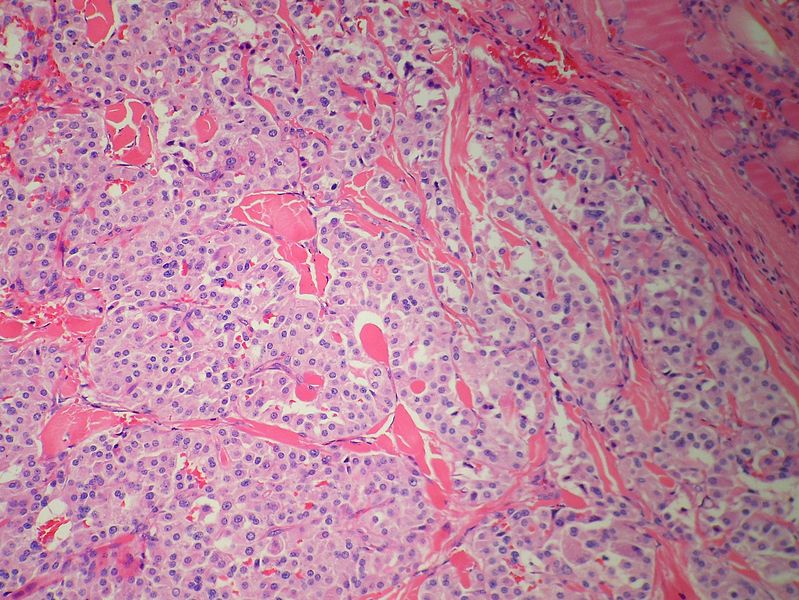
Medullary thyroid carcinoma amyloid<ref> Medullary thyroid cancer librepathology
-
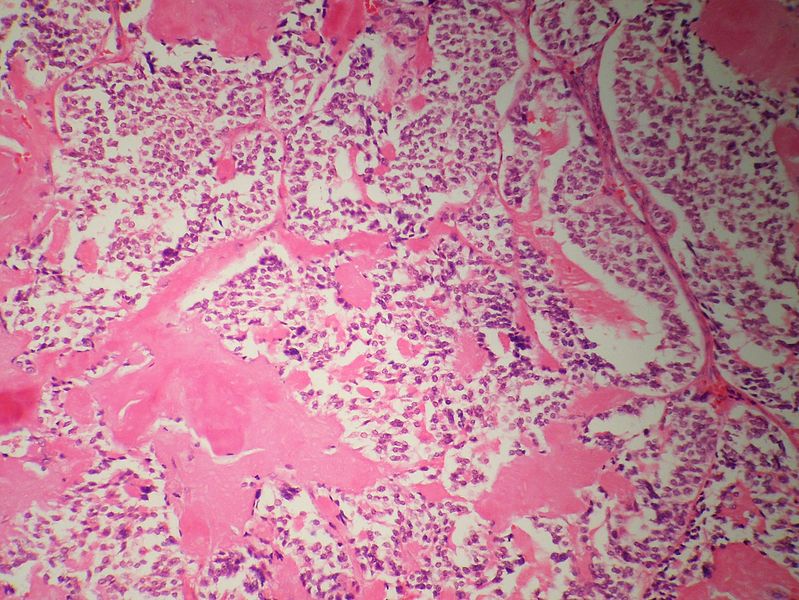
Medullary thyroid carcinoma amyloid<ref> Medullary thyroid cancer librepathology
-
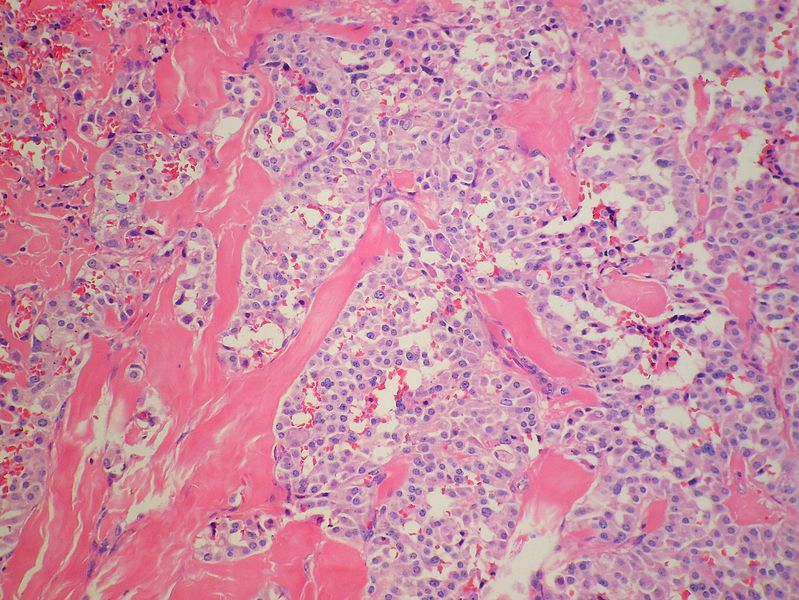
Medullary thyroid carcinoma amyloid<ref> Medullary thyroid cancer librepathology
-
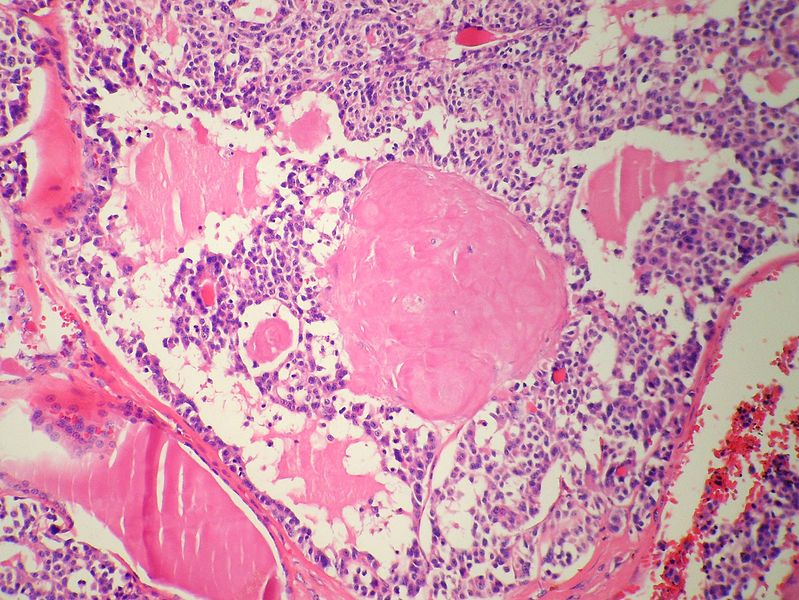
Medullary thyroid carcinoma amyloid<ref> Medullary thyroid cancer librepathology
-
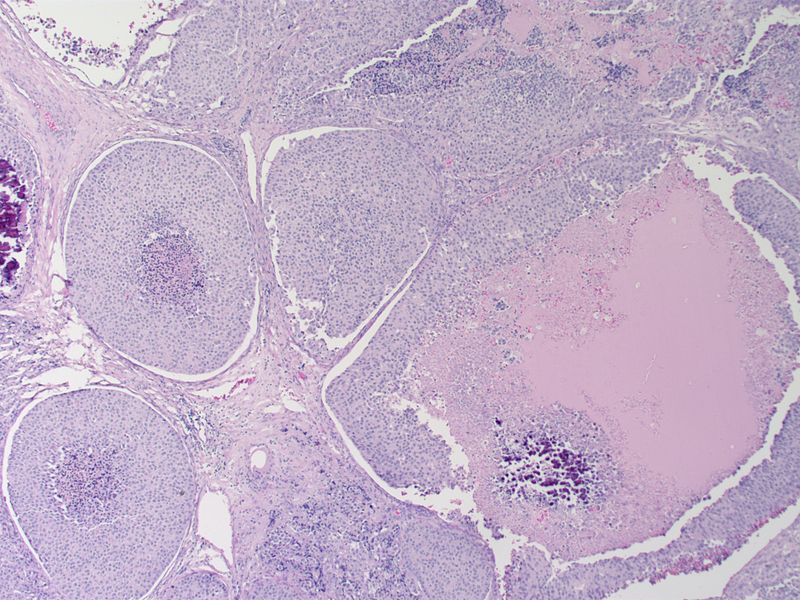
Medullary thyroid carcinoma comedonecrosis<ref> Medullary thyroid cancer librepathology
-
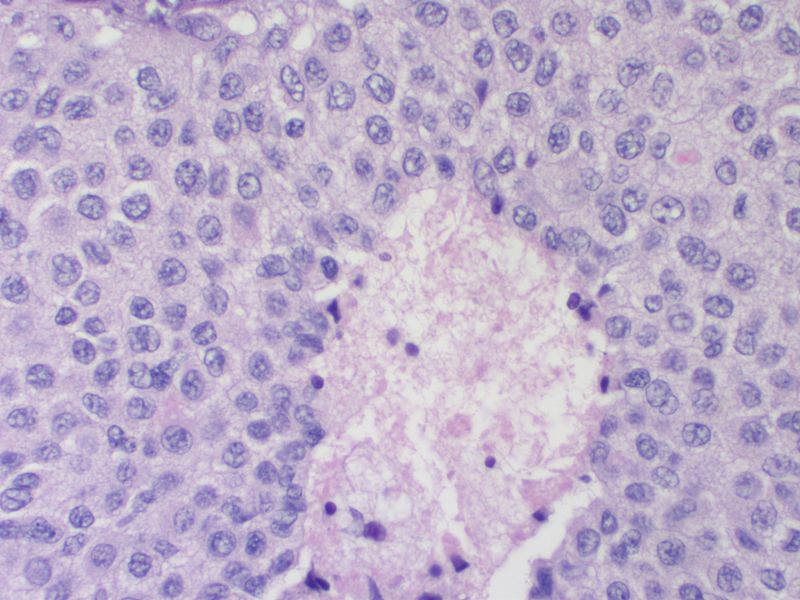
Medullary thyroid carcinoma comedonecrosis<ref> Medullary thyroid cancer librepathology
-

Medullary thyroid carcinoma comedonecrosis<ref> Medullary thyroid cancer librepathology
-
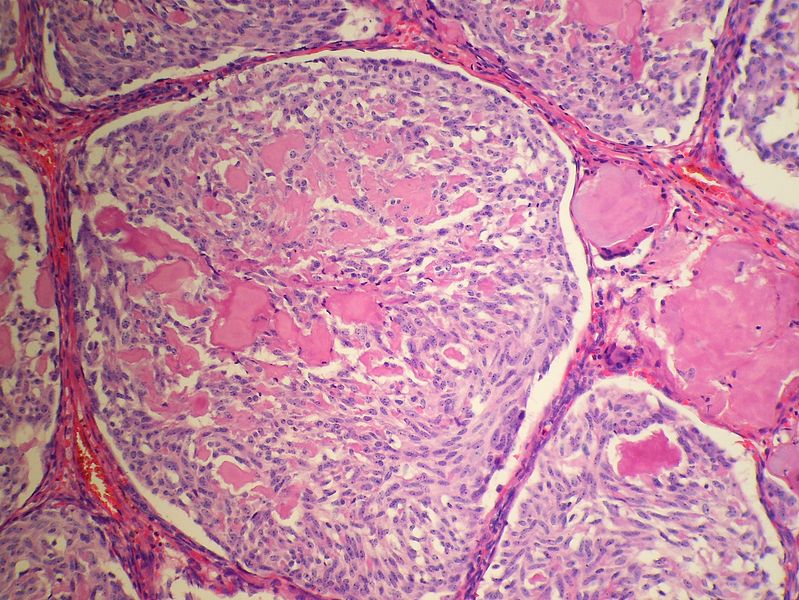
Medullary thyroid carcinoma spindle cell<ref> Medullary thyroid cancer librepathology
-
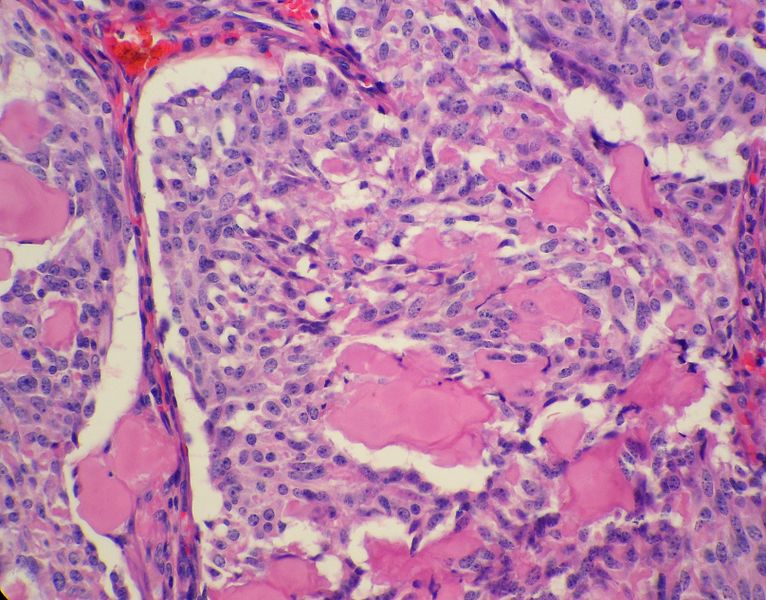
Medullary thyroid carcinoma spindle cell<ref> Medullary thyroid cancer librepathology
-
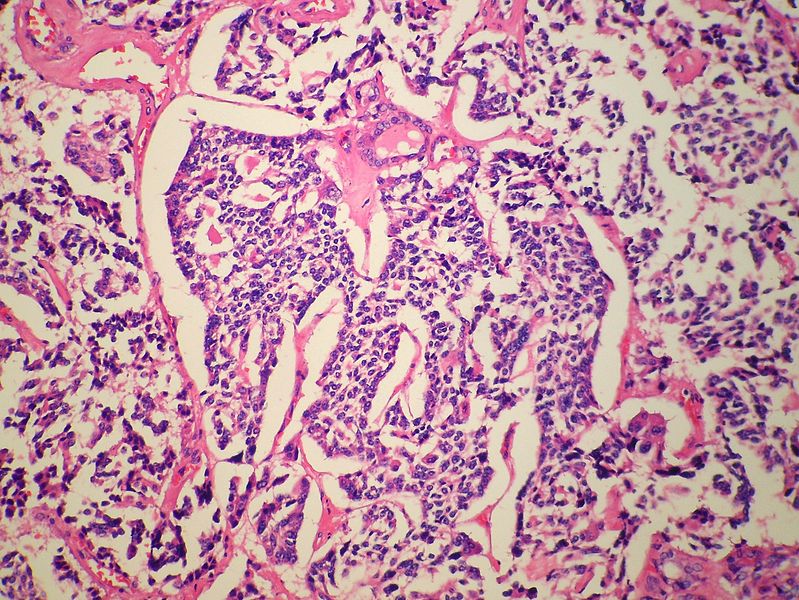
Medullary thyroid carcinoma spindle cell<ref> Medullary thyroid cancer librepathology
-
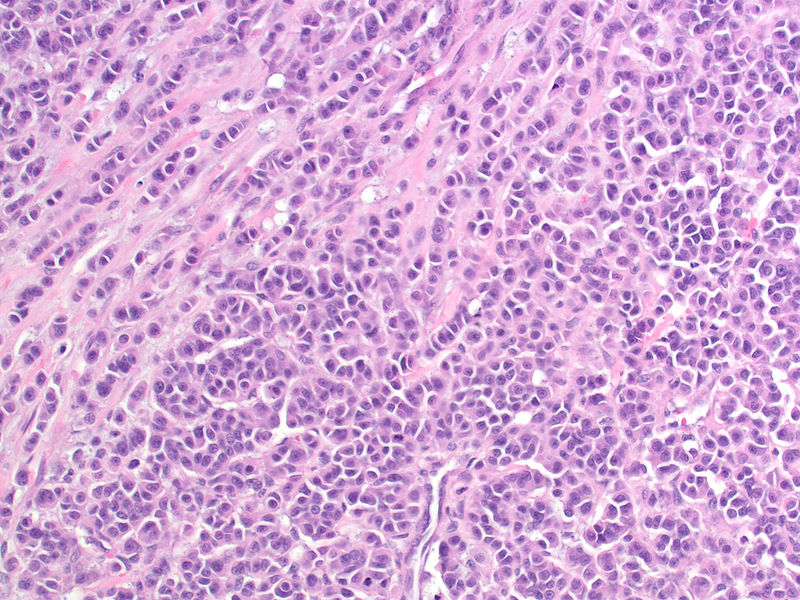
Medullary thyroid carcinoma spindle cell<ref> Medullary thyroid cancer librepathology
-
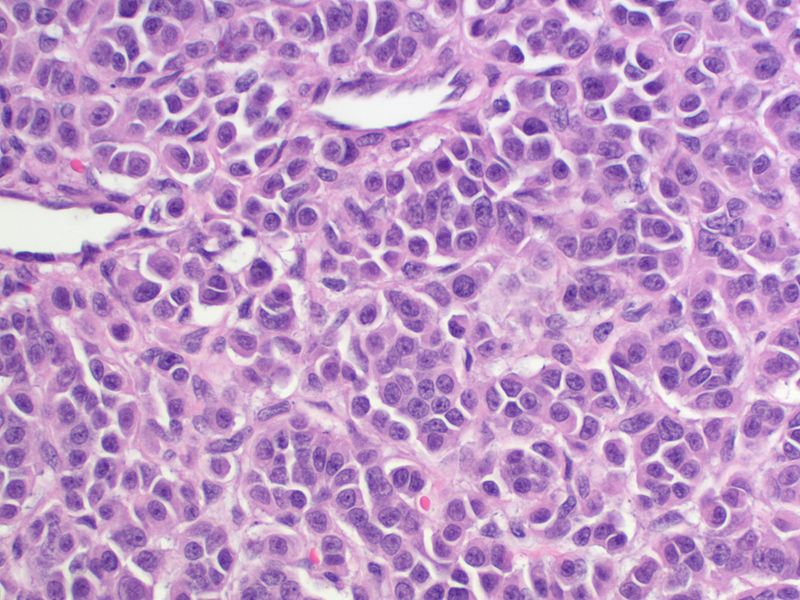
Medullary thyroid carcinoma spindle cell<ref> Medullary thyroid cancer librepathology
-
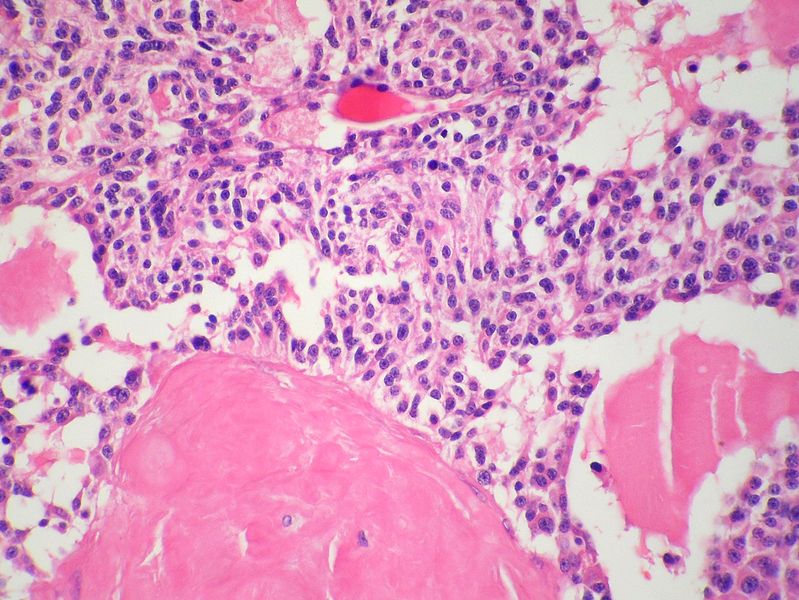
Medullary thyroid carcinoma<ref> Medullary thyroid cancer librepathology
















.jpg)

References
- ↑ Hu MI, Vassilopoulou-Sellin R, Lustig R, Lamont JP. "Thyroid and Parathyroid Cancers" in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.