Spinocerebellar ataxia
| Spinocerebellar ataxia | |
| File:Brain-cerebellum.png | |
|---|---|
| Cerebellum (in blue) of the human brain | |
| ICD-10 | G11 |
| ICD-9 | 334 |
| DiseasesDB | 12339 |
| MeSH | D020754 |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
Spinocerebellar ataxia (SCA) is a genetic disease with multiple types, each of which could be considered a disease in its own right.
Symptoms
Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and often associated with poor coordination of hands, speech, and eye movements. Frequently, atrophy of the cerebellum occurs. [1]
As with other forms of ataxia, SCA results in unsteady and clumsy motion of the body due to a failure of the fine coordination of muscle movements, along with other symptoms.
The symptoms of the condition vary with the specific type (there are several), and with the individual patient. Generally, a person with ataxia retains full mental capacity but may progressively lose physical control.
Treatment and prognosis
There is no known cure for spinocerebellar ataxia, which is a progressive disease (it gets worse with time), although not all types cause equally severe disability.
Treatments are generally limited to softening symptoms, not the disease itself. The condition can be irreversible. A person with this disease will usually end up needing to use a wheelchair, and eventually they may need assistance to perform daily tasks.
The treatment of incoordination or ataxia, then mostly involves the use of adaptive devices to allow the ataxic individual to maintain as much independence as possible. Such devices may include a cane, crutches, walker, or wheelchair for those with impaired gait; devices to assist with writing, feeding, and self care if hand and arm coordination are impaired; and communication devices for those with impaired speech.
Many patients with hereditary or idiopathic forms of ataxia have other symptoms in addition to ataxia. Medications or other therapies might be appropriate for some of these symptoms, which could include tremor, stiffness, depression, spasticity, and sleep disorders, among others.
Both onset of initial symptoms and duration of disease can be subject to variation. If the disease is caused by a polyglutamine trinucleotide repeat CAG expansion, a longer expansion may lead to an earlier onset and a more radical progression of clinical symptoms.
Diagnosis
It can be easily misdiagnosed as another neurological condition, such as multiple sclerosis (MS).
One means of identifying the disease is with an MRI to view the brain. Once the disease has progressed sufficiently, the cerebellum (a part of the brain) can be seen to have visibly shrunk. The most precise means of identifying SCA, including the specific type, is through DNA analysis. Some, but far from all, types of SCA may be inherited, so a DNA test may be done on the children of a sufferer, to see if they are at risk of developing the condition.
SCA is related to olivopontocerebellar atrophy (OPCA); SCA types 1, 2, and 7 are also types of OPCA. However, not all types of OPCA are types of SCA, and vice versa. This overlapping classification system is both confusing and controversial to some in this field.
Types
The following is a list of some, not all, types of Spinocerebellar ataxia. The first ataxia gene was identified in 1993 for a dominantly inherited type. It was called “Spinocerebellar ataxia type 1" (SCA1). Subsequently, as additional dominant genes were found they were called SCA2, SCA3, etc. Usually, the "type" number of "SCA" refers to the order in which the gene was found. At this time, there are at least 29 different gene mutations which have been found (not all listed).
Identifying the different types of SCA now requires knowledge of the normal genetic code, and faults in this code, which are contained in a person's DNA (Deoxyribonucleic acid). The "CAG" mentioned below is one of many three-letter sequences that makes up the genetic code, this specific one coding the amino acid glutamine. Thus, those ataxias with poly CAG expansions, along with several other neurodegenerative diseases resulting from a poly CAG expansion, are referred to as polyglutamine diseases.
| SCA Type | Average Onset (Range in Years) |
Average Duration (Range in Years) |
What the patient experiences | Common origin | Problems with DNA |
|---|---|---|---|---|---|
| SCA1[2] (ATXN1) | 4th decade (<10 to >60) |
15 years (10-28) |
Hypermetric saccades, slow saccades, upper motor neuron (note: saccades relates to eye movement) |
CAG repeat, 6p (Ataxin 1) | |
| SCA2[3] (ATXN2) | 3rd - 4th decade (<10 to >60) |
10 years (1-30) |
Diminished velocity saccades areflexia (absence of neurologic reflexes) |
Cuba | CAG repeat, 12q |
| SCA3[4] (MJD) (ATXN3) | 4th decade (10-70) |
10 years (1-20) |
Also called Machado-Joseph disease (MJD)[5] Gaze-evoked nystagmus (a rapid, involuntary, oscillatory motion of the eyeball) upper motor neuron slow saccades |
Azores (Portugal) |
CAG repeat, 14q |
| SCA4 (PLEKHG4) | 4th - 7th decade (19-72) |
Decades | areflexia (absence of neurologic reflexes) | Chromosome 16q | |
| SCA5 (SPTBN2) | 3rd - 4th decade (10-68) |
>25 years | Pure cerebellar | Chromosome 11 | |
| SCA6[6] (CACNA1A) | 5th - 6th decade (19-71) |
>25 years | Downbeating nystagmus, positional vertigo Symptoms can appear for the first time as late as 65 years old. |
CAG repeat, 19p Calcium channel gene | |
| SCA7[7] (ATXN7) | 3rd - 4th decade (0.5 - 60) |
20 years (1-45; early onset correlates with shorter duration) |
Macular degeneration, upper motor neuron, slow saccades | CAG repeat, 3p (Ataxin 7) | |
| SCA8[8] (IOSCA) | 39 yrs (18-65) |
Normal lifespan | Horizontal nystagmus (a rapid, involuntary, oscillatory motion of the eyeball) | CTG repeat, 13q | |
| SCA10[9] (ATXN10) | 36 years | 9 years | ataxia, seizures | Mexico | Chromosome 22q linked pentanucleotide repeat |
| SCA11 | 30 yrs (15-70) |
Normal lifespan | Mild, remain ambulatory (able to walk about on one's own) | 15q | |
| SCA12[10] (PPP2R2B) | 33 yrs (8-55) |
Head and hand tremor, akinesia (loss of normal motor function, resulting in impaired muscle movement) |
CAG repeat, 5q | ||
| SCA13 | Childhood or adulthood depending on mutation | Depending on KCNC3 (a kind of gene) | Mental retardation | 19q | |
| SCA14[11] (PRKCG) | 28 yrs (12-42) |
Decades (1-30) |
Myoclonus (a sudden twitching of muscles or parts of muscles, without any rhythm or pattern, occurring in various brain disorders) | 19q | |
| SCA16 | 39 yrs (20-66) |
1-40 years | Head and hand tremor | 8q | |
| SCA17 (TBP) | CAG repeat, 6q (TATA-binding protein) | ||||
| SCA19, SCA22 | Mild cerebellar syndrome, dysarthria | ||||
| SCA25 | 1.5-39 yrs | Unknown | ataxia with sensory neuropathy, vomiting and gastrointestinal pain. | 2p |
Others include SCA18, SCA20, SCA21, SCA23, SCA26, SCA28, and SCA29.
Four X-linked types have been described (302500, 302600, 301790, 301840), but only the first of these has so far been tied to a gene (SCAX1).
Inheritance
The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. The hereditary ataxias can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
- Numerous types of autosomal dominant cerebellar ataxias are now known for which specific genetic information is available. Synonyms for autosomal dominant cerebellar ataxias (ADCA) used prior to the current understanding of the molecular genetics were Marie's ataxia, inherited olivopontocerebellar atrophy, cerebello-olivary atrophy, or the more generic term "spinocerebellar degeneration." (Spinocerebellar degeneration is a rare inherited neurological disorder of the central nervous system characterized by the slow degeneration of certain areas of the brain. There are three forms of spinocerebellar degeneration: Types 1, 2, 3. Symptoms begin during adulthood.)
- There are five typical autosomal recessive disorders in which ataxia is a prominent feature: Friedreich ataxia, ataxia-telangiectasia, ataxia with vitamin E deficiency, ataxia with oculomotor apraxia (AOA), spastic ataxia. Disorder Subdivisions: Friedreich's ataxia, Spinocerebellar ataxia, Ataxia telangiectasia, Vasomotor ataxia, Vestibulocerebellar, Ataxiadynamia, Ataxiophemia, Olivopontocerebellar atrophy, and Charcot-Marie-Tooth disease.
-
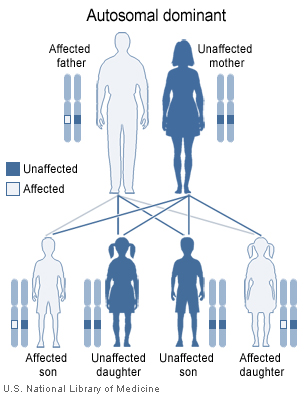 There are numerous types of autosomal dominant cerebellar ataxias
There are numerous types of autosomal dominant cerebellar ataxias -
 There are five typical autosomal recessive disorders in which ataxia is a prominent feature
There are five typical autosomal recessive disorders in which ataxia is a prominent feature


{kind=link}
References
- ↑ Genes and Disease at nlm.nih.gov - Gives a concise description of SCA, along with a picture of shrunken degenerated cerebellum.
- ↑ sca1 at NIH/UW GeneTests
- ↑ sca2 at NIH/UW GeneTests
- ↑ sca3 at NIH/UW GeneTests
- ↑ Template:NINDS
- ↑ sca6 at NIH/UW GeneTests
- ↑ sca7 at NIH/UW GeneTests
- ↑ sca8 at NIH/UW GeneTests
- ↑ sca10 at NIH/UW GeneTests
- ↑ sca12 at NIH/UW GeneTests
- ↑ sca14 at NIH/UW GeneTests
External links
- http://www.ataxia.org - National Ataxia Foundation is dedicated to helping families with ataxia through research, education, and support.
- Cerebellar Degenerations at tchain.com
- http://leedsdna.info/tests/DRPLA.htm
- http://www.ataxiaforums.co.uk
- Ichi Rittoru no Namida (One Litre of Tears) - A Japanese drama based on the true story of a girl who suffered from Spinocerebellar ataxia.
- CureAtaxia.org - One Chicago family's fight against SCA Type 1 Ataxia
- OMIM: 183090 Template:RareDiseases
- OMIM: 164400 Template:RareDiseases
- OMIM: 607317 Template:RareDiseases
- OMIM: 600224 Template:RareDiseases
- OMIM: 605259 Template:RareDiseases
- OMIM: 117360 Template:RareDiseases
- OMIM: 608703 Template:RareDiseases
- OMIM: 600223 Template:RareDiseases
- OMIM: 606002 Template:RareDiseases
- OMIM: 271250 Template:RareDiseases
- OMIM: 609306 Template:RareDiseases
- OMIM: 608029 Template:RareDiseases
- OMIM: 607346 Template:RareDiseases
- OMIM: 608687 Template:RareDiseases
- OMIM: 302600 Template:RareDiseases
- OMIM: 606937 Template:RareDiseases
- OMIM: 301840 Template:RareDiseases
- OMIM: 301790 Template:RareDiseases
- OMIM: 109150 Template:RareDiseases
- OMIM: 607458 Template:RareDiseases
- Template:RareDiseases
- OMIM: 164500 Template:RareDiseases
- OMIM: 607454 Template:RareDiseases
- OMIM: 610245 Template:RareDiseases
- OMIM: 610246 Template:RareDiseases
- OMIM: 603680 Template:RareDiseases
- OMIM: 607250 Template:RareDiseases
- OMIM: 605361 Template:RareDiseases
- OMIM: 271245 Template:RareDiseases
- Template:RareDiseases
- OMIM: 301310 Template:RareDiseases
- OMIM: 229300 Template:RareDiseases
Template:Diseases of the nervous system Template:Trinucleotide repeat disorders
de:Spinozerebelläre Ataxie it:Atassia spinocerebellare th:สไปโนซีรีเบลลาร์อะแท็กเซีย