Idursulfase
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Rabin Bista, M.B.B.S. [2]
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Black Box Warning
|
WARNING:
See full prescribing information for complete Boxed Warning.
RISK OF ANAPHYLAXIS:
|
Overview
Idursulfase is a hydrolytic lysosomal glycosaminoglycan (GAG)-specific enzyme that is FDA approved for the treatment of Hunter syndrome (Mucopolysaccharidosis II, MPS II). There is a Black Box Warning for this drug as shown here. Common adverse reactions include headache, pruritus, musculoskeletal pain, urticaria, diarrhea, cough, pyrexia, rash, vomiting.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Indications
- ELAPRASE is indicated for patients with Hunter syndrome (Mucopolysaccharidosis II, MPS II). ELAPRASE has been shown to improve walking capacity in patients 5 years and older.
- In patients 16 months to 5 years of age, no data are available to demonstrate improvement in disease-related symptoms or long term clinical outcome; however, treatment with ELAPRASE has reduced spleen volume similarly to that of adults and children 5 years of age and older.
- The safety and efficacy of ELAPRASE have not been established in pediatric patients less than 16 months of age
Dosage
- The recommended dosage regimen of ELAPRASE is 0.5 mg per kg of body weight administered once weekly as an intravenous infusion.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
DOSAGE FORMS AND STRENGTHS
- Injection: 6 mg/3 mL (2 mg/mL) in single-use vials
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Idursulfase in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Idursulfase in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
Indications
- ELAPRASE is indicated for patients with Hunter syndrome (Mucopolysaccharidosis II, MPS II). ELAPRASE has been shown to improve walking capacity in patients 5 years and older.
- In patients 16 months to 5 years of age, no data are available to demonstrate improvement in disease-related symptoms or long term clinical outcome; however, treatment with ELAPRASE has reduced spleen volume similarly to that of adults and children 5 years of age and older.
- The safety and efficacy of ELAPRASE have not been established in pediatric patients less than 16 months of age
Dosage
- The recommended dosage regimen of ELAPRASE is 0.5 mg per kg of body weight administered once weekly as an intravenous infusion.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Idursulfase in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Idursulfase in pediatric patients.
Contraindications
- None
Warnings
|
WARNING:
See full prescribing information for complete Boxed Warning.
RISK OF ANAPHYLAXIS:
|
Hypersensitivity Reactions Including Anaphylaxis
- Serious hypersensitivity reactions, including anaphylaxis, have occurred during and up to 24 hours after infusion. Some of these reactions were life-threatening and included respiratory distress, hypoxia, hypotension, urticaria, and angioedema of the throat or tongue, regardless of duration of the course of treatment.
- If anaphylactic or other acute reactions occur, immediately discontinue the infusion of ELAPRASE and initiate appropriate medical treatment. When severe reactions have occurred during clinical trials, subsequent infusions were managed with antihistamine and/or corticosteroids prior to or during infusions, a slower rate of ELAPRASE infusion, and/or early discontinuation of the ELAPRASE infusion.
- In clinical trials with ELAPRASE, 16 of 108 (15%) patients experienced hypersensitivity reactions during 26 of 8,274 infusions (0.3%) that involved adverse events in at least two of the following three body systems: cutaneous, respiratory, or cardiovascular. Of these 16 patients, 11 experienced anaphylactic reactions during 19 of 8,274 infusions (0.2%) with symptoms of bronchospasm, cyanosis, dyspnea, erythema, edema (facial and peripheral), flushing, rash, respiratory distress, urticaria, vomiting, and wheezing.
- In postmarketing reports, patients receiving ELAPRASE experienced anaphylactic reactions up to several years after initiating treatment. Some patients were reported to have repeated anaphylactic events over a two- to four-month time period. Medical management included treatment with antihistamines, inhaled beta-adrenergic agonists, corticosteroids, oxygen, and vasopressors. Treatment was discontinued for some patients, while others continued treatment with premedication and a slower infusion rate.
- Due to the potential for severe reactions, appropriate medical support should be readily available when ELAPRASE is administered. Observe patients closely for an appropriate period of time after administration of ELAPRASE, taking into account the time to onset of anaphylaxis seen in premarketing clinical trials and postmarketing reports. Inform patients of the signs and symptoms of anaphylaxis, and instruct them to seek immediate medical care should signs and symptoms occur.
Risk of Hypersensitivity, Serious Adverse Reactions, and Antibody Development in Hunter Syndrome Patients with Severe Genetic Mutations
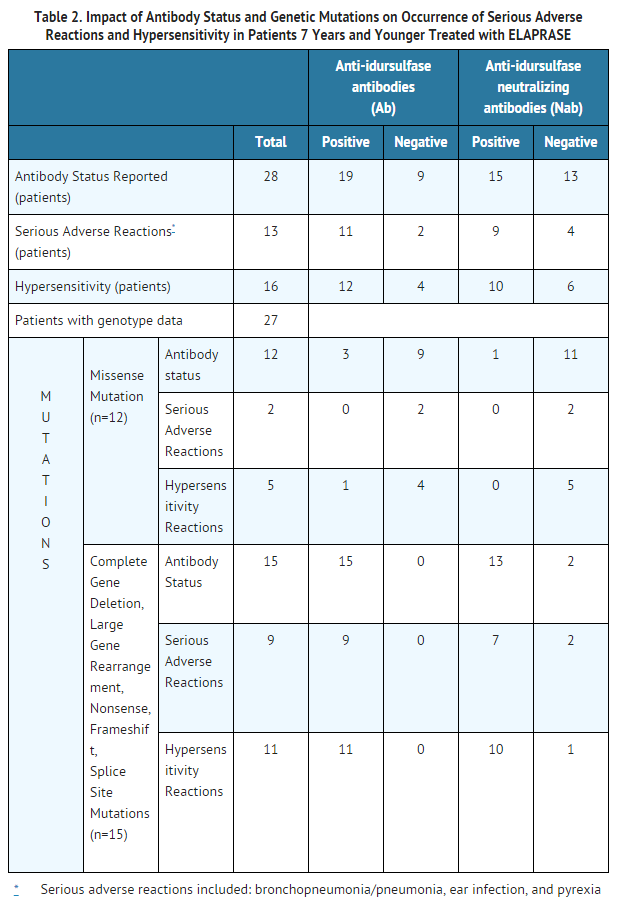
- In the clinical trial of Hunter syndrome patients aged 7 years and younger, patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations experienced a higher incidence of hypersensitivity reactions, serious adverse reactions and anti-idursulfase antibody development than Hunter syndrome patients with missense mutations. Eleven of 15 (73%) patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations and five of 12 (42%) patients with missense mutations experienced hypersensitivity reactions. Nine of 15 (60%) patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations and two of 12 (17%) patients with missense mutations had serious adverse reactions. All 15 patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations developed anti-idursulfase (ELAPRASE) antibodies, compared to only 3 patients with missense mutations (Table 2). Thirteen patients with these mutations developed neutralizing antibodies, which interfere with ELAPRASE uptake into the cell or ELAPRASE enzyme activity, compared to only one patient with missense mutation.
Risk of Acute Respiratory Complications
- Patients with compromised respiratory function or acute febrile or respiratory illness at the time of ELAPRASE infusion may be at higher risk of life-threatening complications from hypersensitivity reactions. Careful consideration should be given to the patient's clinical status prior to administration of ELAPRASE and consider delaying the ELAPRASE infusion. One patient with a tracheostomy, severe airway disease and acute febrile illness experienced respiratory distress, hypoxia, cyanosis, and seizure with a loss of consciousness during ELAPRASE infusion.
Risk of Acute Cardiorespiratory Failure
- Caution should be exercised when administering ELAPRASE to patients susceptible to fluid overload, or patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function for whom fluid restriction is indicated. These patients may be at risk of serious exacerbation of their cardiac or respiratory status during infusions. Appropriate medical support and monitoring measures should be readily available during ELAPRASE infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient
Adverse Reactions
Clinical Trials Experience
Clinical Trials Experience
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
- The following serious adverse reactions are described below and elsewhere in the labeling:
- Hypersensitivity Reactions Including Anaphylaxis
- In clinical trials, the most common adverse reactions (>10%) following ELAPRASE treatment were hypersensitivity reactions, and included rash, urticaria, pruritus, flushing, pyrexia, and headache. Most hypersensitivity reactions requiring intervention were ameliorated with slowing of the infusion rate, temporarily stopping the infusion, with or without administering additional treatments including antihistamines, corticosteroids or both prior to or during infusions.
- In clinical trials, the most frequent serious adverse reactions following ELAPRASE treatment were hypoxic episodes. Other notable serious adverse reactions that occurred in the ELAPRASE-treated patients but not in the placebo-treated patients included one case each of: cardiac arrhythmia, pulmonary embolism, cyanosis, respiratory failure, infection, and arthralgia.
Clinical Trials in Patients 5 Years and Older
- A 53-week, double-blind, placebo-controlled clinical trial of ELAPRASE was conducted in 96 male patients with Hunter syndrome, ages 5-31 years old. Of the 96 patients, 83% were White, non-Hispanic. Patients were randomized to three treatment groups, each with 32 patients: ELAPRASE 0.5 mg/kg once weekly, ELAPRASE 0.5 mg/kg every other week, or placebo. Hypersensitivity reactions were reported in 69% (22 of 32) of patients who received once-weekly treatment of ELAPRASE.
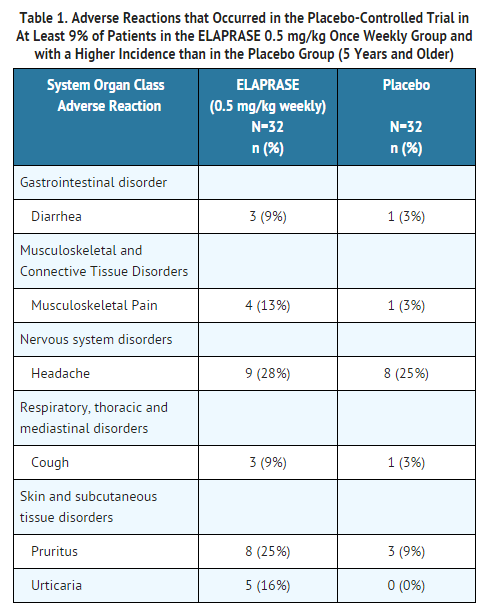
- Table 1 summarizes the adverse reactions that occurred in at least 9% of patients (≥3 patients) in the ELAPRASE 0.5 mg/kg once weekly group and with a higher incidence than in the placebo group.
This image is provided by the National Library of Medicine.

- Additional adverse reactions that occurred in at least 9% of patients (≥3 patients) in the ELAPRASE 0.5 mg/kg every other week group and with a higher incidence than in the placebo group included: rash (19%), flushing (16%), fatigue (13%), tachycardia (9%), and chills (9%).
Extension Trial
- An open-label extension trial was conducted in patients who completed the placebo-controlled trial. Ninety-four of the 96 patients who were enrolled in the placebo-controlled trial consented to participate in the extension trial. All 94 patients received ELAPRASE 0.5 mg/kg once weekly for 24 months. No new serious adverse reactions were reported. Approximately half (53%) of patients experienced hypersensitivity reactions during the 24-month extension trial. In addition to the adverse reactions listed in Table 1, common hypersensitivity reactions occurring in at least 5% of patients (≥ 5 patients) in the extension trial included: rash (23%), pyrexia (9%), flushing (7%), erythema (7%), nausea (5%), dizziness (5%), vomiting (5%), and hypotension (5%).
Clinical Trial in Patients 7 Years and Younger
- A 53-week, open-label, single-arm, safety trial of once weekly ELAPRASE 0.5 mg/kg treatment was conducted in patients with Hunter syndrome, ages 16 months to 4 years old (n=20) and ages 5 to 7.5 years old (n=8) at enrollment. Patients experienced similar adverse reactions as those observed in clinical trials in patients 5 years and older, with the most common adverse reactions following ELAPRASE treatment being hypersensitivity reactions (57%). A higher incidence of the following common hypersensitivity reactions were reported in this younger age group: pyrexia (36%), rash (32%) and vomiting (14%). The most common serious adverse reactions occurring in at least 10% of patients (≥ 3 patients) included: bronchopneumonia/pneumonia (18%), ear infection (11%), and pyrexia (11%).
- Twenty-seven patients had results of genotype analysis: 15 patients had complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations and 12 patients had missense mutations.
- Safety results demonstrated that patients with complete gene deletion, large gene rearrangement, nonsense, frameshift, or splice site mutations are more likely to experience hypersensitivity reactions and have serious adverse reactions following ELAPRASE administration, compared to patients with missense mutations. Table 2 summarizes these findings.
This image is provided by the National Library of Medicine.

Immunogenicity
Clinical Trials in Patients 5 Years and Older
- As with all therapeutic proteins, there is potential for immunogenicity. In clinical trials in patients 5 years and older, 63 of the 64 patients treated with ELAPRASE 0.5 mg/kg once weekly or placebo for 53 weeks, followed by ELAPRASE 0.5 mg/kg once weekly in the extension trial, had immunogenicity data available for analysis. Of the 63 patients, 32 (51%) patients tested positive for anti-idursulfase IgG antibodies (Ab) at least one time (Table 2). Of the 32 Ab-positive patients, 23 (72%) tested positive for Ab at three or more different time points (persistent Ab). The incidence of hypersensitivity reactions was higher in patients who tested positive for Ab than those who tested negative.
- Thirteen of 32 (41%) Ab-positive patients also tested positive for antibodies that neutralize idursulfase uptake into cells (uptake neutralizing antibodies, uptake NAb) or enzymatic activity (activity NAb) at least one time, and 8 (25%) of Ab-positive patients had persistent NAb. There was no clear relationship between the presence of either Ab or NAb and therapeutic response.
Clinical Trial in Patients 7 Years and Younger
- In the clinical trial in patients 7 years and younger, 19 of 28 (68%) patients treated with ELAPRASE 0.5 mg/kg once weekly tested Ab-positive. Of the 19 Ab-positive patients, 16 (84%) tested positive for Ab at three or more different time points (persistent Ab). In addition, 15 of 19 (79%) Ab-positive patients tested positive for NAb, with 14 of 15 (93%) NAb-positive patients having persistent NAb.
- All 15 patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations tested positive for Ab (Table 2). Of these 15 patients, neutralizing antibodies were observed in 13 (87%) patients. The NAbs in these patients developed earlier (most reported to be positive at Week 9 rather than at Week 27, as reported in clinical trials in patients older than 5 years of age) and were associated with higher titers and greater in vitro neutralizing activity than in patients older than 5 years of age. The presence of Ab was associated with reduced systemic idursulfase exposure.
- The immunogenicity data reflect the percentage of patients whose test results were positive for antibodies to idursulfase in specific assays, and are highly dependent on the sensitivity and specificity of these assays. The observed incidence of positive antibody in an assay may be influenced by several factors, including sample handling, timing of sample collection, concomitant medication, and underlying disease. For these reasons, comparison of the incidence of antibodies to idursulfase with the incidence of antibodies to other products may be misleading.
Postmarketing Experience
- The following adverse reactions have been identified during post approval use of ELAPRASE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- In post-marketing experience, late-emergent symptoms and signs of anaphylactic reactions have occurred up to 24 hours after initial treatment and recovery from an initial anaphylactic reaction. In addition, patients experienced repeated anaphylaxis over a two- to four-month period, up to several years after initiating ELAPRASE treatment.
- A seven year-old male patient with Hunter syndrome, who received ELAPRASE at twice the recommended dosage (1 mg/kg weekly) for 1.5 years, experienced two anaphylactic events after 4.5 years of treatment. Treatment has been withdrawn.
- Serious adverse reactions that resulted in death included cardiorespiratory arrest, respiratory failure, respiratory distress, cardiac failure, and pneumonia.
Drug Interactions
There is limited information regarding Idursulfase Drug Interactions in the drug label.
Use in Specific Populations
Pregnancy
- Teratogenicity studies have not been conducted with ELAPRASE. A pre- and postnatal development study in rats showed no evidence of adverse effects on pre- and postnatal development at intravenous doses up to 12.5 mg/kg, administered twice weekly (about 4 times the recommended human weekly dose of 0.5 mg/kg based on body surface area). There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Idursulfase in women who are pregnant.
Labor and Delivery
There is no FDA guidance on use of Idursulfase during labor and delivery.
Nursing Mothers
- ELAPRASE was excreted in breast milk of lactating rats at a concentration higher (4 to 5-fold) than that of the plasma. It is not known whether ELAPRASE is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ELAPRASE is administered to a nursing woman.
Pediatric Use
- Clinical trials with ELAPRASE were conducted in 96 patients with Hunter syndrome, ages 5 to 31 years old, with the majority of the patients in the pediatric age group (median age 15 years old). In addition, an open-label, uncontrolled clinical trial was conducted in 28 patients with Hunter syndrome, ages 16 months to 7.5 years old. Patients 16 months to 5 years of age demonstrated reduction in spleen volume that was similar to that of adults and children 5 years and older. However, there are no data to support improvement in disease-related symptoms or long term clinical outcome in patients 16 months to 5 years of age.
The safety and effectiveness of ELAPRASE have not been established in pediatric patients less than 16 months of age.
Geriatic Use
- Clinical studies of ELAPRASE did not include patients older than 31 years of age. It is not known whether older patients respond differently from younger patients.
Gender
There is no FDA guidance on the use of Idursulfase with respect to specific gender populations.
Race
There is no FDA guidance on the use of Idursulfase with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Idursulfase in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Idursulfase in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Idursulfase in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Idursulfase in patients who are immunocompromised.
Administration and Monitoring
Administration
Preparation Instructions
- Prepare and use ELAPRASE according to the following steps using aseptic technique:
- Determine the total volume of ELAPRASE to be administered and the number of vials needed based on the patient's weight and the recommended dose of 0.5 mg/kg.

- Round up to the next whole vial to determine the total number of vials needed. Remove the required number of vials from the refrigerator to allow them to reach room temperature.
- Before withdrawing the ELAPRASE solution from the vial, visually inspect each vial for particulate matter and discoloration. The ELAPRASE solution should be clear to slightly opalescent and colorless. Do not use if the solution is discolored or if there is particulate matter in the solution. Do not shake the ELAPRASE solution.
- Withdraw the calculated volume of ELAPRASE from the appropriate number of vials.
- Add the calculated volume of ELAPRASE solution to a 100 mL bag of 0.9% Sodium Chloride Injection, USP for intravenous infusion.
- Mix gently. Do not shake the solution.
Administration Instructions
- Administer the diluted ELAPRASE solution to patients using a low-protein-binding infusion set equipped with a low-protein-binding 0.2 micrometer (µm) in-line filter.
- The total volume of infusion should be administered over a period of 3 hours, which may be gradually reduced to 1 hour if no hypersensitivity reactions are observed. Patients may require longer infusion times if hypersensitivity reactions occur; however, infusion times should not exceed 8 hours. The initial infusion rate should be 8 mL per hour for the first 15 minutes. If the infusion is well tolerated, the rate of infusion may be increased by 8 mL per hour increments every 15 minutes. The infusion rate should not exceed 100 mL per hour. The infusion rate may be slowed, temporarily stopped, or discontinued for that visit in the event of hypersensitivity reactions. ELAPRASE should not be infused with other products in the infusion tubing.
Storage and Stability
- ELAPRASE does not contain preservatives; therefore, after dilution with saline, the infusion bags should be used immediately. If immediate use is not possible, the diluted solution should be stored refrigerated at 2°C to 8°C (36°F to 46 °F) for up to 24 hours. Other than during infusion, do not store the diluted ELAPRASE solution at room temperature. Any unused product or waste material should be discarded and disposed of in accordance with local requirements.
Monitoring
Caution should be exercised when administering ELAPRASE to patients susceptible to fluid overload, or patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function for whom fluid restriction is indicated. These patients may be at risk of serious exacerbation of their cardiac or respiratory status during infusions. Appropriate medical support and monitoring measures should be readily available during ELAPRASE infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient
IV Compatibility
There is limited information regarding IV Compatibility of Idursulfase in the drug label.
Overdosage
- One patient with Hunter syndrome, who received ELAPRASE at twice the recommended dosage for one and a half years, experienced two anaphylactic reactions over a 3-month period 4.5 years after initiating ELAPRASE treatment.
Pharmacology
Idursulfase
| |
| Systematic (IUPAC) name | |
| ? | |
| Identifiers | |
| CAS number | |
| ATC code | A16 |
| PubChem | ? |
| DrugBank | |
| Chemical data | |
| Formula | ? |
| Mol. mass | ? |
| Pharmacokinetic data | |
| Bioavailability | ? |
| Metabolism | ? |
| Half life | ? |
| Excretion | ? |
| Therapeutic considerations | |
| Pregnancy cat. |
? |
| Legal status | |
| Routes | ? |
Mechanism of Action
- Hunter syndrome (Mucopolysaccharidosis II, MPS II) is an X-linked recessive disease caused by insufficient levels of the lysosomal enzyme iduronate-2-sulfatase. This enzyme cleaves the terminal 2-O-sulfate moieties from the glycosaminoglycans (GAG) dermatan sulfate and heparan sulfate. Due to the missing or defective iduronate-2-sulfatase enzyme in patients with Hunter syndrome, GAG progressively accumulate in the lysosomes of a variety of cells, leading to cellular engorgement, organomegaly, tissue destruction, and organ system dysfunction.
- ELAPRASE is intended to provide exogenous enzyme for uptake into cellular lysosomes. Mannose-6-phosphate (M6P) residues on the oligosaccharide chains allow binding of the enzyme to the M6P receptors on the cell surface, leading to cellular internalization of the enzyme, targeting to intracellular lysosomes and subsequent catabolism of accumulated GAG.
Structure
- ELAPRASE is a formulation of idursulfase, a purified form of human iduronate-2-sulfatase, a lysosomal enzyme. Idursulfase is produced by recombinant DNA technology in a human cell line. Idursulfase is an enzyme that hydrolyzes the 2-sulfate esters of terminal iduronate sulfate residues from the glycosaminoglycans dermatan sulfate and heparan sulfate in the lysosomes of various cell types.
- Idursulfase is a 525-amino acid glycoprotein with a molecular weight of approximately 76 kilodaltons. The enzyme contains eight asparagine-linked glycosylation sites occupied by complex oligosaccharide structures. The enzyme activity of idursulfase is dependent on the post-translational modification of a specific cysteine to formylglycine. Idursulfase has a specific activity ranging from 46 to 74 units/mg of protein (one unit is defined as the amount of enzyme required to hydrolyze 1 µmole of heparin disaccharide substrate per hour under the specified assay conditions).
- ELAPRASE is administered as an intravenous infusion and supplied as a sterile, nonpyrogenic clear to slightly opalescent, colorless solution that must be diluted prior to administration in 0.9% Sodium Chloride Injection, USP. Each vial contains an extractable volume of 3 mL with an idursulfase concentration of 2 mg/mL at a pH of approximately 6. Each vial contains 6 mg idursulfase, sodium chloride (24 mg), sodium phosphate monobasic monohydrate (6.75 mg), sodium phosphate dibasic heptahydrate (2.97 mg), and polysorbate 20 (0.66 mg). ELAPRASE does not contain preservatives. Each vial is for single use only.
Pharmacodynamics
- Decreases in urinary GAG levels were observed following treatment with ELAPRASE. The responsiveness of urinary GAG to dosage alterations of ELAPRASE is unknown, and the relationship of urinary GAG to other measures of clinical response has not been established. Patients who tested positive for anti-idursulfase antibodies (Ab) experienced a less pronounced decrease in urinary GAG levels
Pharmacokinetics
Clinical Trials in Patients 5 Years and Older
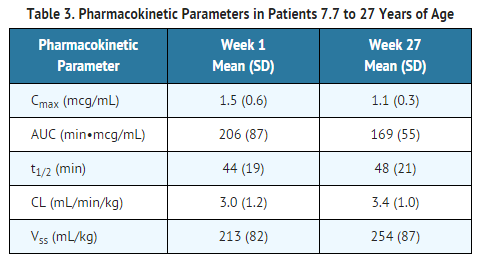
- The pharmacokinetic characteristics of idursulfase were evaluated in 59 patients with Hunter syndrome. The serum concentration of idursulfase was quantified using an antigen-specific ELISA assay. The area under the concentration-time curve (AUC) increased in a greater than dose proportional manner as the dose increased from 0.15 mg/kg to 1.5 mg/kg following a single 1-hour infusion of ELAPRASE. The pharmacokinetic parameters at the recommended dose regimen (0.5 mg/kg ELAPRASE administered weekly as a 3-hour infusion) were determined at Week 1 and Week 27 in 10 patients 7.7 to 27 years of age (Table 3). There were no apparent differences in PK parameter values between Week 1 and Week 27 regardless of the antibody status in these patients.
This image is provided by the National Library of Medicine.

Clinical Trial in Patients 7 Years and Younger
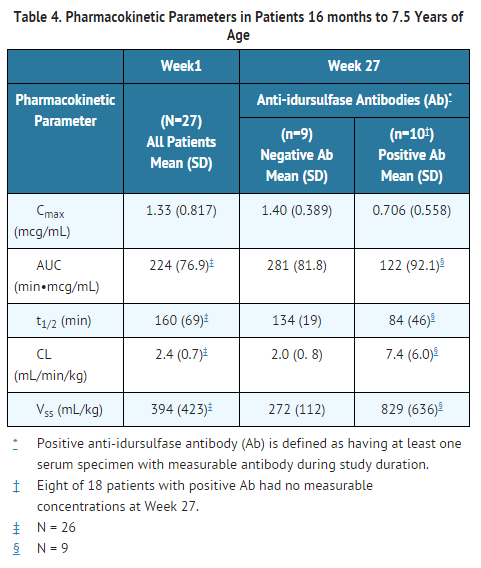
- Idursulfase pharmacokinetics was evaluated in 27 patients with Hunter syndrome 16 months to 7.5 years of age who received ELAPRASE 0.5 mg/kg once weekly as a 3-hour infusion. The presence of anti-idursulfase antibody (Ab) was associated with a reduced systemic exposure of idursulfase. Eight of the 18 Ab-positive patients had no measurable idursulfase concentrations. An additional 9 Ab-positive patients had decreased Cmax, AUC, and t1/2 at Week 27 compared to Week 1 (Table 4). Idursulfase pharmacokinetics was similar between Week 1 and Week 27 in Ab-negative patients (Table 4).
This image is provided by the National Library of Medicine.

- All patients with the complete gene deletion or large gene rearrangement genotype (n = 8) developed Ab at Week 27. Five of these eight patients had no measurable idursulfase concentrations at Week 27, and three had a lower systemic exposure at Week 27 compared to Week 1.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment of Fertility
- Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with ELAPRASE.
- ELAPRASE at intravenous doses up to 5 mg/kg administered twice weekly (about 1.6 times the recommended human weekly dose based on body surface area) had no effect on fertility and reproductive performance in male rats.
Clinical Studies
Clinical Trials in Patients 5 Years and Older
- The safety and efficacy of ELAPRASE were evaluated in a 53-week, randomized, double-blind, placebo-controlled clinical trial of 96 patients with Hunter syndrome. The trial included patients with deficiency in iduronate-2-sulfatase enzyme activity and a percent predicted forced vital capacity (% predicted FVC) less than 80%. The age of patients ranged from 5 to 31 years. Patients received ELAPRASE 0.5 mg/kg once per week (n=32), ELAPRASE 0.5 mg/kg once every other week (n=32), or placebo (n=32).
- The primary efficacy outcome assessment was a two-component composite score based on the sum of the ranks of the change from baseline to Week 53 in distance walked in six minutes (6-minute walk test) and the ranks of the change in % predicted FVC. This two-component composite primary endpoint differed statistically significantly between the three groups, and the difference was greatest between the placebo group and the once weekly treatment group (once weekly ELAPRASE vs. placebo, p=0.0049).
- Examination of the individual components of the composite score showed that, in the adjusted analysis, the weekly ELAPRASE-treated group experienced a 35 meter greater mean increase in the distance walked in six minutes compared to placebo. The changes in %-predicted FVC were not statistically significant (Table 5).
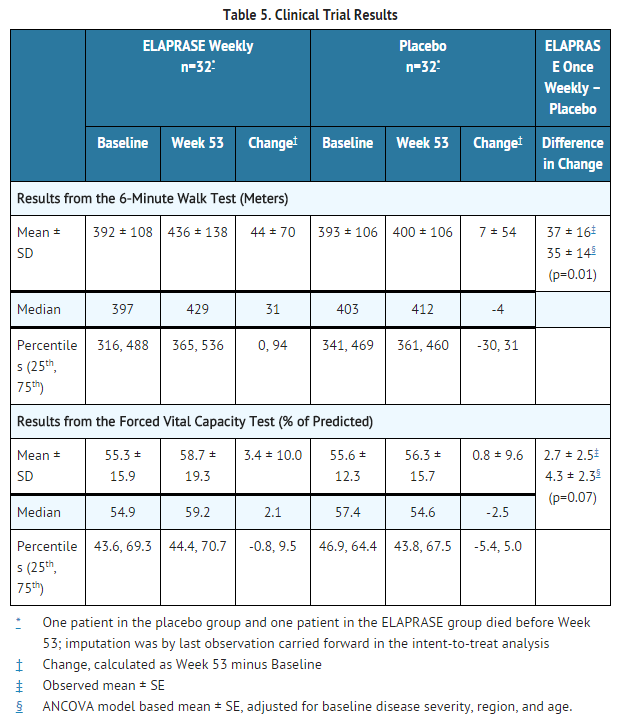
This image is provided by the National Library of Medicine.

- Pharmacodynamic assessments included urinary GAG levels and changes in liver and spleen size. Urinary GAG levels were elevated in all patients at baseline. Following 53 weeks of treatment, mean urinary GAG levels were reduced in the ELAPRASE once weekly group, although GAG levels still remained above the upper limit of normal in half of the ELAPRASE-treated patients. Urinary GAG levels remained elevated and essentially unchanged in the placebo group. Sustained reductions in both liver and spleen volumes were observed in the ELAPRASE once weekly group through Week 53 compared to placebo. There were essentially no changes in liver and spleen volumes in the placebo group.
Extension Trial
- Patients who participated in the placebo-controlled trial were eligible to continue treatment in an open-label extension trial. During the extension trial, all patients received ELAPRASE 0.5mg/kg once weekly for 24 months.
- Patients who were treated with ELAPRASE once weekly and every other week in the placebo-controlled trial demonstrated improvement in distance walked in the 6-minute walk test for an additional 8 months of treatment in the extension trial. There was no change in mean %-predicted FVC in all Hunter syndrome patients after 6 months of treatment in the extension trial; however, a slight decrease in mean %-predicted FVC was demonstrated through to month 24 of the extension trial. The long-term effect of ELAPRASE on pulmonary function in Hunter syndrome patients is unclear.
- There were no further reductions in mean urinary GAG levels in patients initially treated with ELAPRASE once weekly; however, the patients treated with ELAPRASE every other week during the placebo-controlled trial experienced further reductions in mean urinary GAG levels after changing to a more frequent dosing regimen during the extension trial. The persistence of reduced urinary GAG levels did not correlate with the long term effect demonstrated by the 6-minute walk test distance or %-predicted FVC.
Clinical Trial in Patients 7 Years and Younger
- A 53-week, open-label, multicenter, single-arm trial was conducted to assess the safety, pharmacokinetics, and pharmacodynamics of ELAPRASE 0.5 mg/kg once weekly in male Hunter syndrome patients aged 7 years and younger. Safety results demonstrated that patients with complete gene deletion or large gene rearrangement mutations are more likely to develop antibodies, including neutralizing antibodies, and to experience hypersensitivity reactions with ELAPRASE administration. In patients who remained antibody negative, the pharmacokinetic profile, reduction in urinary GAG excretion levels, and reduction in spleen volume were similar to those of adults and children 5 years and older. In patients who were persistently antibody positive, the presence of anti-idursulfase antibody was associated with reduced systemic exposure of idursulfase and a less pronounced decrease in urinary GAG levels
How Supplied
- ELAPRASE is supplied as a sterile injection in a 5 mL Type I glass vial. The vials are closed with a butyl rubber stopper with fluororesin coating and an aluminum overseal with a blue flip-off plastic cap.
- Each carton contains a single vial NDC 54092-700-01
Storage
- Store ELAPRASE vials in the carton at 2°C to 8°C (36°F to 46°F) to protect from light. Do not freeze or shake. Do not use ELAPRASE after the expiration date on the vial.
Images
Drug Images
{{#ask: Page Name::Idursulfase |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 6 mg/3 mL Vial Carton
elaprase® (idursulfase) injection
6 mg/3 mL (2 mg/mL)
Single Use Vial, Discard Unused Portion
Must be diluted for intravenous infusion
Shire
Rx Only
This image is provided by the National Library of Medicine.

Ingredients and Appearance
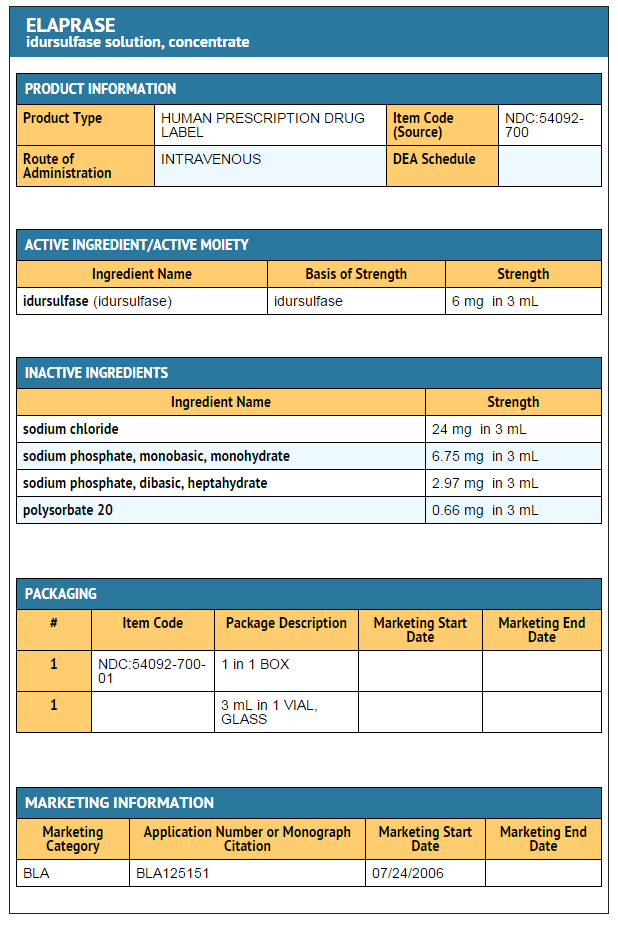
This image is provided by the National Library of Medicine.

{{#ask: Label Page::Idursulfase |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- Patients should be advised that life-threatening anaphylactic reactions have occurred in some patients during and up to 24 hours after ELAPRASE therapy. Patients who have experienced anaphylactic reactions may require prolonged observation. Patients with compromised respiratory function or acute respiratory disease may be at risk of serious acute exacerbation of their respiratory compromise due to hypersensitivity reactions.
- A Hunter Outcome Survey has been established in order to understand better the variability and progression of Hunter syndrome (MPS II) in the population as a whole, and to monitor and evaluate long-term treatment effects of ELAPRASE. Patients and their physicians are encouraged to participate in this program. For more information, call Shire Human Genetic Therapies, Inc. at 1-866-888-0660.
Precautions with Alcohol
- Alcohol-Idursulfase interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- ELAPRASE®[1]
Look-Alike Drug Names
There is limited information regarding Idursulfase Look-Alike Drug Names in the drug label.
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
{{#subobject:
|Page Name=Idursulfase
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}