Complement system
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]

The complement system is a biochemical cascade which helps clear pathogens from an organism. It is part of the larger immune system that is not adaptable and does not change over the course of an individual's lifetime; as such it belongs to the innate immune system. However, it can be recruited and brought into action by the adaptive immune system.
The complement system consists of a number of small proteins found in the blood, normally circulating as inactive zymogens. When stimulated by one of several triggers, proteases in the system cleave specific proteins to release cytokines and initiate an amplifying cascade of further cleavages. The end result of this activation cascade is massive amplification of the response and activation of the cell-killing membrane attack complex. Over 20 proteins and protein fragments make up the complement system, including serum proteins, serosal proteins, and cell membrane receptors. These proteins are synthesized mainly in the liver, and they account for about 5% of the globulin fraction of blood serum.
Three biochemical pathways activate the complement system: the classical complement pathway, the alternative complement pathway, and the mannose-binding lectin pathway.[1]
History
In the late 19th century, blood serum was found to contain a "factor" or "principle" which was capable of killing bacteria. In 1896, Jules Bordet, a young Belgian scientist in Paris at the Pasteur Institute, demonstrated that this principle could be analyzed into two components: a heat-stable and a heat-labile component. (Heat-labile meaning that it lost its effectiveness if the serum was heated.) The heat-stable component was found to confer immunity against specific microorganisms, while the heat-labile component was found to be responsible for the non-specific antimicrobial activity conferred by all normal serum. This heat-labile component is what we now call "complement".
The term "complement" was introduced by Paul Ehrlich in the late 1890s, as part of his larger theory of the immune system. According to this theory, the immune system consists of cells which have specific receptors on their surface to recognize antigens. Upon immunization with an antigen, more of these receptors are formed, and they are then shed from the cells to circulate in the blood. These receptors, which we now call "antibodies", were called by Ehrlich "amboceptors" to emphasize their bifunctional binding capacity: they recognize and bind to a specific antigen, but they also recognize and bind to the heat-labile antimicrobial component of fresh serum. Ehrlich therefore named this heat-labile component "complement", because it is something in the blood which "complements" the cells of the immune system.
Ehrlich believed that each antigen-specific amboceptor had its own specific complement, while Bordet believed that there is only one type of complement. In the early 20th century, this controversy was resolved when it was understood that complement can act in combination with specific antibodies, or on its own in a non-specific way.
Overview

The three pathways all generate homologous variants of the protease C3-convertase. The classical complement pathway typically requires antibodies for activation (specific immune response), while the alternative and mannose-binding lectin pathways can be activated by C3 hydrolysis or antigens without the presence of antibodies (non-specific immune response). In all three pathways, a C3-convertase cleaves and activates component C3, creating C3a and C3b and causing a cascade of further cleavage and activation events. C3b binds to the surface of pathogens leading to greater internalization by phagocytic cells by opsonization. C5a is an important chemotactic protein, helping recruit inflammatory cells. Both C3a and C5a have anaphylatoxin activity, directly triggering degranulation of mast cells as well as increasing vascular permeability and smooth muscle contraction. C5b initiates the membrane attack pathway, which results in the membrane attack complex (MAC), consisting of C5b, C6, C7, C8, and polymeric C9.[2] MAC is the cytolytic endproduct of the complement cascade; it forms a transmembrane channel, which causes osmotic lysis of the target cell. Kupffer cells and other macrophage cell types help clear complement-coated pathogens. As part of the innate immune system, elements of the complement cascade can be found in species earlier than vertebrates; most recently in the protostome horseshoe crab species, putting the origins of the system back further than was previously thought.
Classical pathway

The classical pathway is triggered by activation of the C1-complex (which consists of one molecule C1q and two molecules C1r and C1s), either by C1q's binding to antibodies from classes M and G, complexed with antigens, or by its binding C1q to the surface of the pathogen. This binding leads to conformational changes in C1q molecule, which leads to the activation of two C1r (serine protease) molecules. Then they cleave C1s (another serine protease). The C1-complex now binds to and splits C2 and C4, producing C2b (used to be called C2a, but nomenclature has been updated because "a's" are conventionally smaller) and C4b. The inhibition of C1r and C1s is controlled by C1-inhibitor. C4b and C2b bind to form C3-convertase (C4b2b complex: Many older texts, however, use the traditional nomenclature for this, calling it C4b2a). Production of C3-convertase leads to cleavage of C3 into C3a and C3b; the latter joins with C2b and C4b (the C3 convertase) to make C5 convertase.
Alternative pathway
The alternative pathway is triggered by C3 hydrolysis directly on the surface of a pathogen. It does not rely on a pathogen-binding protein like the other pathways.[1] In the alternative pathway, the protein C3 is produced in the liver, and is then cleaved into C3a and C3b by enzymes in the blood. If there is no pathogen in the blood, the C3a and C3b protein fragments will be deactivated. However, if there is a nearby pathogen, some of the C3b is bound to the plasma membrane of the pathogen. Then, it will bind to factor B. This complex will then be cleaved by factor D into Ba and the alternative pathway C3-convertase, Bb.
The C3bBb complex, which is "hooked" onto the surface of the pathogen, will then act like a "chain saw", catalyzing the hydrolysis of C3 in the blood into C3a and C3b, which positively effects the number of C3bBb hooked onto a pathogen.
After hydrolysis of C3, C3b complexes to become C3bBbC3b, which cleaves C5 into C5a and C5b. C5a and C3a are known to trigger mast cell degranulation. C5b with C6, C7, C8, and C9 (C5b6789) complex to form the membrane attack complex, also known as MAC, which is inserted into the cell membrane, "punches a hole", and initiates cells lysis.
Lectin pathway (MBL - MASP)
The lectin pathway is homologous to the classical pathway, but with the opsonin, mannan-binding lectin (MBL) and ficolins, instead of C1q. This pathway is activated by binding mannan-binding lectin to mannose residues on the pathogen surface, which activates the MBL-associated serine proteases, MASP-1, MASP-2, MASP-3, which can then split C4 into C4a and C4b and C2 into C2a and C2b. C4b and C2a then bind together to form C3-convertase, as in the classical pathway. Ficolins are homologous to MBL and function via MASP in a similar way. In invertebrates without an adaptive immune system, ficolins are expanded and their binding specificities diversified to compensate for the lack of pathogen-specific recognition molecules.
Regulation of the Complement System
The complement system has the potential to be extremely damaging to host tissues meaning its activation must be tightly regulated. The complement system is regulated by complement control proteins, which are present at a higher concentration in the blood plasma than the complement proteins themselves. Some complement control proteins are present on the membranes of self-cells preventing them from being targeted by complement. One example is CD59, which inhibits C9 polymerisation during the formation of the membrane attack complex.
Role in disease
It is thought that the complement system might play a role in many diseases with an immune component, such as Barraquer-Simons Syndrome, asthma, lupus erythematosus, glomerulonephritis, various forms of arthritis, autoimmune heart disease, multiple sclerosis, inflammatory bowel disease, and ischemia-reperfusion injuries. The complement system is also becoming increasingly implicated in diseases of the central nervous system such as Alzheimer's disease, and other neurodegenerative conditions.
Deficiencies of the terminal pathway predispose to both autoimmune disease and infections (particularly meningitis, due to the role that the C56789 complex plays in attacking Gram negative bacteria).
Mutations in the complement regulators factor H and membrane cofactor protein have been associated with atypical haemolytic uraemic syndrome.[3][4] Moreover a common single nucleotide polymorphism in factor H (Y402H) has been associated with the common eye disease 'age related macular degeneration'.[5] Both of these disorders are currently thought to be due to aberrant complement activation on host surfaces.
Modulation by infections
Recent research has suggested that the complement system is manipulated during HIV/AIDS to further damage the body.[6][7]
Additional images
-
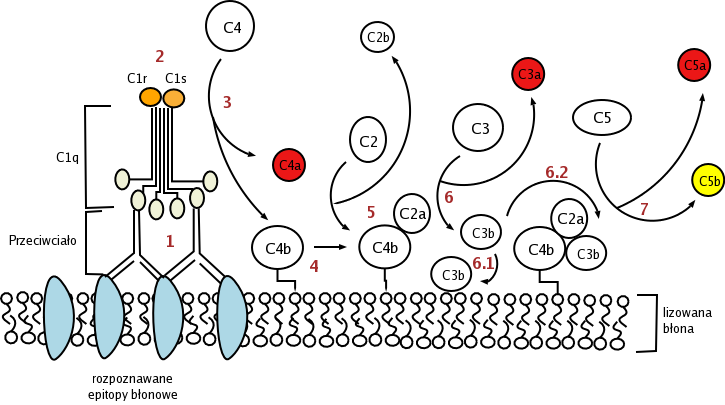 Classical
Classical -
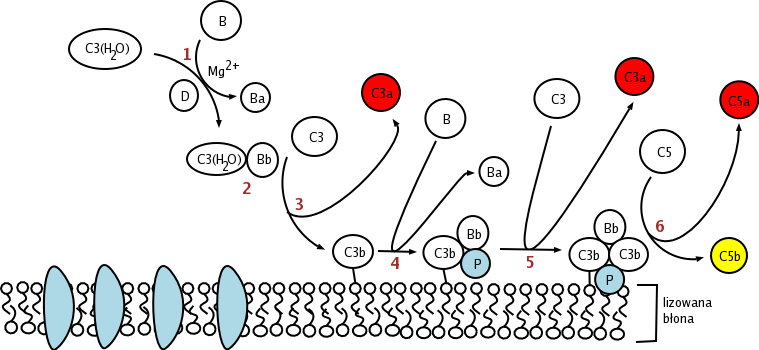 Alternative
Alternative -
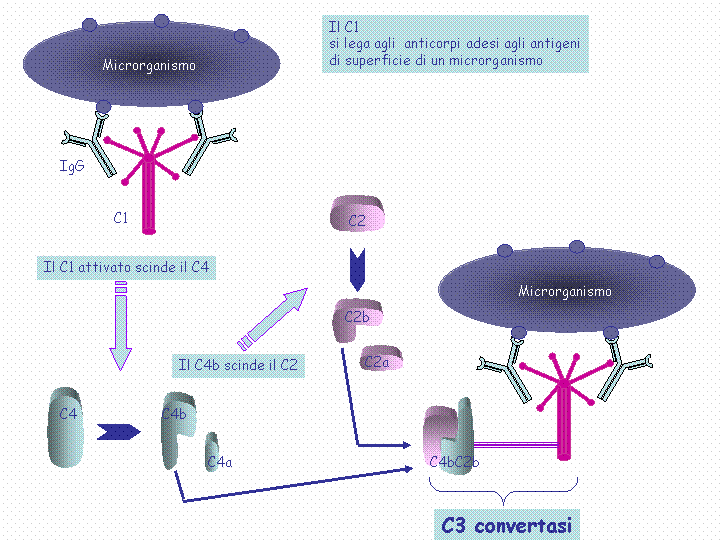 Classical
Classical



{kind=link}
References
- ↑ 1.0 1.1 Janeway CA Jr., Travers P, Walport M, Shlomchik MJ (2001). Immunobiology (5th ed. ed.). Garland Publishing. (via NCBI Bookshelf) ISBN 0-8153-3642-X.
- ↑ Goldman AS, Prabhakar BS (1996). The Complement System. in: Baron's Medical Microbiology (Baron S et al, eds.) (4th ed. ed.). Univ of Texas Medical Branch. (via NCBI Bookshelf) ISBN 0-9631172-1-1.
- ↑ Dragon-Durey MA, Frémeaux-Bacchi V (2005). "Atypical haemolytic uraemic syndrome and mutations in complement regulator genes". Springer Semin. Immunopathol. 27 (3): 359–74. doi:10.1007/s00281-005-0003-2. PMID 16189652.
- ↑ Zipfel PF, Misselwitz J, Licht C, Skerka C (2006). "The role of defective complement control in hemolytic uremic syndrome". Semin. Thromb. Hemost. 32 (2): 146–54. doi:10.1055/s-2006-939770. PMID 16575689.
- ↑ Mooijaart SP, Koeijvoets KM, Sijbrands EJ, Daha MR, Westendorp RG (2007). "Complement Factor H polymorphism Y402H associates with inflammation, visual acuity, and cardiovascular mortality in the elderly population at large". doi:10.1016/j.exger.2007.08.001. PMID 17869048.
- ↑ Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, Wright JR, Complement Levels and Activity in the Normal and LPS-Injured Lung, American Journal of Physiology: Lung Cellular and Molecular Physiology. 2006 Oct 27; PMID 17071722
- ↑ Datta PK, Rappaport J, HIV and Complement: Hijacking an immune defence, Biomedicine and Pharmacotherapy, 2006 Nov; 60(9):561-568 PMID 16978830
cs:Komplement (biologie) de:Komplementsystem ia:Complimento it:Sistema del complemento he:מערכת המשלים nl:Complementsysteem simple:Complement fi:Komplementti (biologia) sv:Komplement (biologi)