Multiple myeloma pathophysiology
|
Multiple myeloma Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Multiple myeloma pathophysiology On the Web |
|
American Roentgen Ray Society Images of Multiple myeloma pathophysiology |
|
Risk calculators and risk factors for Multiple myeloma pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] Associate Editor(s)-in-Chief: Haytham Allaham, M.D. [2] Shyam Patel [3]
Overview
Multiple myeloma arises from post-germinal center plasma cells that are normally involved in production of human immunoglobulins.[1][2][3] Both sporadic events and familial contributions contribute to the pathogenesis of multiple myeloma. Renal involvement by multiple myeloma is catergorized into three entities: light chain cast nephropathy, monoclonal immunoglobulin deposition disease, and amyloidosis. Osseous involvement by multiple myeloma is based on cytokine and cellular interactions that lead to bone breakdown. On microscopic histopathological analysis, abundant eosinophilic cytoplasm, eccentrically placed nucleus, and Russell bodies are characteristic findings of multiple myeloma.[4]
Pathophysiology
Normal physiology of plasma cells
In order to understand the pathophysiology of multiple myeloma, one must understand the normal physiological function of plasma cells. Normally, the proliferation of B lymphocytes and plasma cells is tightly regulated. B lymphocytes normally differentiate into plasma cells. Plasma cells secrete antibodies which function in humoral immunity. Plasma cells are typically polyclonal and can respond to a variety of antigens, which helps combat infection. Under normal circumstances, there is no monoclonality amongst the plasma cell population in a person. Normal plasma cells have normal karyotypes and have no mutations. These plasma cells are functionally intact in their ability to contribute to humoral immunity.
Pathogenesis
- Sporadic events: The pathophysiology of multiple myeloma is based on aberrant plasma cell proliferation due to a carcinogenic insult. The malignant cells arise from post-germinal center plasma cells. Upon chromosome and/or gene damage, such as rearrangements or point mutations, the normal regulatory mechanisms that govern B lymphocyte and plasma cell proliferation are lost. Often, an immunoglobulin heavy chain-encoding gene (such as the IgH gene) translocates to another chromosome and inserts itself upstream of a cell cycle control gene. Since IgH is normally expressed at high levels in plasma cells, the translocation of IgH to other sites can drive autonomous cell proliferation and result in cancer. This genetic change results in dysregulation of the oncogene which is thought to be an important initiating event in the pathogenesis of multiple myeloma. Multiple myeloma arises from post-germinal center plasma cells that are normally involved in production of human immunoglobulins. The genes involved in the pathogenesis of multiple myeloma include the heavy chain gene (on chromosome 14, locus 14q32) and oncogenes (often located on chromosomes 4, 6, 11, 16, and 20).[5]
- Familial predisposition: A familial predisposition to myeloma exists. Hyperphosphorylation of the paratarg proteins, a tendency which is inherited in an autosomal dominant manner, appears to be a common mechanism in these families. This tendency is more common in African American patients with myeloma and may contribute to the higher rates of myeloma in this group.[2][6]
Pathophysiology of renal involvement
Abnormal antibody fragments are produced in multiple myeloma and are deposited in various organs, such as the kidneys. There are three major forms of renal damage in patients with multiple myeloma.
- Cast nephropathy: End-organ damage to the kidneys is typically due to light chain cast nephropathy. The pathophysiology of this type of renal involvement is based on light chain deposition in the renal tubules, which results in obstruction. Free light chains are readily filtered at the glomerulus and are reabsorbed in the proximal tubule of the nephron. This reabsorption occurs via the megalin-cubulin transport system.[7] In patients with multiple myeloma, there is excess production of free light chains, and the ability of the nephron to resorb light chains in the promixal tubule cannot meet the demands of the freely filtered light chains. This results in excess secretion of free light chains in the urine (known as Bence-Jones protein). Eosinophilic proteinaceous casts and crystalline structures can be seen. Cast formation occurs in the tubules due to excess abundance of free light chains that interact with Tamm-Horsfall proteins in the thick ascending loope of Henle.[7] Tubular obstruction ensues, triggering local inflammation which results in interstitial nephritis and fibrosis.[7] The onset of cast nephropathy can be very quick, requiring prompt treatment. Risk factors for development of cast nephropathy include monoclonal immunoglobulin secretion of >10 g/day, sepsis, and volume depletion.[8] Patients can also develop Fanconi syndrome, resulting in dysfunctional reabsorption ability by the proximal tubule, and type II renal tubular acidosis.
- Monoclonal immunoglobulin deposition disease (MIDD): In this subtype of renal involvement by multiple myeloma, the initial pathophysiological process is filtration of monoclonal immunoglobulins and subsequent deposition of immunoglobulins along the tubular or glomerular basement membrane.[8] Deposits of immunoglobulin can have a similar appearance as Kimmelstein-Wilson lesions (seen in diabetes). The immunoglobulins can appear fibroblast-like.
- Light chain amyloidosis: The pathophysiology of renal involvement by light chain amyloidosis begins with beta-pleated sheet formation in the tubules or glomeruli. Beta-pleated sheets form as a result of electrostatic interactions between heparan sulfate proteoglycan and amyloid proteins. Amyloid fibrils usually consist of immunoglobulin light chains (usually lambda light chain) and deposit in the basement membrane. The size of the fibrils vary from 7 to 10 nanometers. A diagnosis of this type of renal involvement is made by the visualization of apple green birefringence upon Congo red staining of the renal specimen.[8] It is frequently associated with nephrotic range proteinuria, in which greater than 3 grams of protein is excreted daily.
Pathophysiology of osseous involvement
The pathophysiology of bony involvement of multiple myeloma involves cytokines and cellular interactions.
- Cytokines: Multiple myeloma is one of the most common malignancies that creates lytic bony lesions. Other cancers that can create lytic bony lesions include renal cell carcinoma, lung cancer, breast cancer, thyroid cancer, and lymphoma. Lytic destruction has a distinct pathophysiology compared to the blastic destruction that is seen in prostate cancer. The pathophysiology of bony disease in multiple myeloma begins with stimulation of osteoclast production and suppression of osteoblast production. The steady state of bone metabolism is shifted in the direction of bone resorption. The molecular mechanism that governs osteoclast activation in multiple myeloma involves nuclear factor kappa B (NFkB), interleukin-3 (IL-3), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-a), and CXCL12, also known as stromal cell-derived factor 1 (SDF1).[9]
- Cellular interactions: At the cellular level, bone breakdown is governed by interactions between multiple myeloma cells, osteoclasts, osteoblasts, mesenchymal stem cells, T lymphocytes and dendritic cells. Multiple myeloma cells chemoattract hematopoietic cells such as dendritic cells, which produce osteoclastic factors that contribute to bone breakdown. Dendritic cells can also transdifferentiate into osteoclasts.[9] Multiple myeloma cells suppress osteoblast production by inhibiting expression of the transcription factor RUNX2, which is critical for osteoblast activity. The immune microenvironment thus plays a major role in the formation of lytic lesions in patients with multiple myeloma. Malignant plasma cells infiltrate hematopoietic sites such as the red bone marrow where they interfere with the production of normal blood cells.[1][2][3][10] The distribution of multiple myeloma mirrors that of red bone marrow in older individuals, and thus multiple myeloma is mostly encountered in the axial skeleton and proximal appendicular skeleton such as:[1]
- Vertebrae (most common)
- Ribs
- Skull
- Shoulder girdle
- Pelvis
- Long bones
- Extra skeletal structures (extraosseous myeloma) (rare)
Gross Pathology
-

Vertebrae in multiple myeloma
(Image courtesy of Melih Aktan M.D.) -
Calvarium in multiple myeloma.
(Image courtesy of Melih Aktan M.D.)


Microscopic Pathology
On microscopic histopathological analysis, multiple myeloma is characterized by the following:[4]
- Abundant eosinophilic cytoplasm
- Eccentrically placed nucleus
- Clock face morphology of the nucleus due to chromatin clumps around the edges
- Russell bodies which are eosinophilic, large (10-15 micrometres), homogenous immunoglobulin-containing inclusions
- Dutcher bodies which are PAS stain +ve intranuclear crystalline rods
- Shown below is a series of microscopic images seen in multiple myeloma:
-
![Multiple Myeloma slide showing plasma cells with large nucleus and scant cytoplasm [11]](/images/a/a5/Multiple_Myeloma.jpg)
Multiple Myeloma slide showing plasma cells with large nucleus and scant cytoplasm [11]
-
Bone marrow aspiration in multiple myeloma.
(Image courtesy of Melih Aktan M.D.) -
Bone marrow biopsy in multiple myeloma.
(Image courtesy of Melih Aktan M.D.) -
Bone marrow in multiple myeloma.
(Image courtesy of Melih Aktan M.D.) -
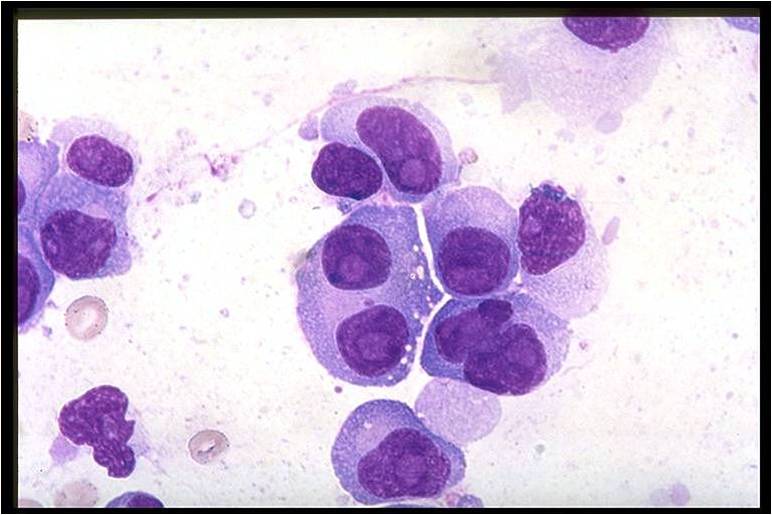
Bone marrow in multiple myeloma.
(Image courtesy of Melih Aktan M.D.) -
![Multiple myeloma slide with intermediate magnification[4]](/images/4/40/Multiple_myeloma_intermed_mag.jpg)
Multiple myeloma slide with intermediate magnification[4]
-
![Multiple myeloma slide with high magnification[4]](/images/d/d5/Multiple_myeloma.jpg)
Multiple myeloma slide with high magnification[4]
-
![Multiple myeloma slide with russell bodies[4]](/images/a/ad/Russell_bodies.jpg)
Multiple myeloma slide with russell bodies[4]
![Multiple Myeloma slide showing plasma cells with large nucleus and scant cytoplasm [11]](/index.php/File:Multiple_Myeloma.jpg)




![Multiple myeloma slide with intermediate magnification[4]](/index.php/File:Multiple_myeloma_intermed_mag.jpg)
![Multiple myeloma slide with high magnification[4]](/index.php/File:Multiple_myeloma.jpg)
![Multiple myeloma slide with russell bodies[4]](/index.php/File:Russell_bodies.jpg)
References
- ↑ 1.0 1.1 1.2 Multiple myeloma. Radiopaedia (2015)http://radiopaedia.org/articles/multiple-myeloma-1 Accessed on September, 20th 2015
- ↑ 2.0 2.1 2.2 Multiple myeloma. Wikipedia (2015)https://en.wikipedia.org/wiki/Multiple_myeloma#Pathophysiology Accessed on September, 20th 2015
- ↑ 3.0 3.1 Multiple myeloma. Medlineplus (2015)https://www.nlm.nih.gov/medlineplus/multiplemyeloma.html Accessed on September, 20th 2015
- ↑ 4.0 4.1 4.2 4.3 4.4 Multiple myeloma. Librepathology (2015)http://www.wikidoc.org/index.php?title=Multiple_myeloma_pathophysiology&action=edit§ion=1 Accessed on September, 20th 2015
- ↑ Kyle RA, Rajkumar SV (2004). "Multiple myeloma". N Engl J Med. 351 (18): 1860–73. doi:10.1056/NEJMra041875. PMID 15509819.
- ↑ Koura DT, Langston AA (2013). "Inherited predisposition to multiple myeloma". Ther Adv Hematol. 4 (4): 291–7. doi:10.1177/2040620713485375. PMC 3734900. PMID 23926460.
- ↑ 7.0 7.1 7.2 Finkel KW, Cohen EP, Shirali A, Abudayyeh A, American Society of Nephrology Onco-Nephrology Forum (2016). "Paraprotein-Related Kidney Disease: Evaluation and Treatment of Myeloma Cast Nephropathy". Clin J Am Soc Nephrol. 11 (12): 2273–2279. doi:10.2215/CJN.01640216. PMC 5142056. PMID 27526708.
- ↑ 8.0 8.1 8.2 Heher EC, Rennke HG, Laubach JP, Richardson PG (2013). "Kidney disease and multiple myeloma". Clin J Am Soc Nephrol. 8 (11): 2007–17. doi:10.2215/CJN.12231212. PMC 3817918. PMID 23868898.
- ↑ 9.0 9.1 Yaccoby S (2010). "Advances in the understanding of myeloma bone disease and tumour growth". Br J Haematol. 149 (3): 311–21. doi:10.1111/j.1365-2141.2010.08141.x. PMC 2864366. PMID 20230410.
- ↑ What is multiple myeloma. Canadian Cancer Society (2015) http://www.cancer.ca/en/cancer-information/cancer-type/multiple-myeloma/multiple-myeloma/?region=mb Accessed on September, 20th 2015
- ↑ http://picasaweb.google.com/mcmumbi/USMLEIIImages