Hemophilia: Difference between revisions
No edit summary |
No edit summary |
||
| Line 12: | Line 12: | ||
| OMIM_mult = {{OMIM2|306900}} {{OMIM2|264900}} | | OMIM_mult = {{OMIM2|306900}} {{OMIM2|264900}} | ||
| MedlinePlus = 000537 | | MedlinePlus = 000537 | ||
| MeshID = D025861 | | MeshID = D025861 | ||
}} | }} | ||
| Line 158: | Line 156: | ||
==References== | ==References== | ||
{{reflist|2}} | {{reflist|2}} | ||
{{Hematology}} | {{Hematology}} | ||
| Line 207: | Line 190: | ||
[[zh:血友病]] | [[zh:血友病]] | ||
[[Category:Disease state]] | |||
[[Category:Hematology]] | [[Category:Hematology]] | ||
[[Category:Genetic disorders]] | [[Category:Genetic disorders]] | ||
[[Category:Coagulation system]] | |||
[[Category:Mature chapter]] | |||
{{WikiDoc Help Menu}} | {{WikiDoc Help Menu}} | ||
{{WikiDoc Sources}} | {{WikiDoc Sources}} | ||
Revision as of 15:51, 29 July 2011
| Hemophilia | |
 | |
|---|---|
| 17-year-old hemophiliac has a pseudotumor of the calcaneus that expands the bone and has stimulated dense new bone formation. Image courtesy of Professor Peter Anderson DVM PhD and published with permission © PEIR, University of Alabama at Birmingham, Department of Pathology | |
| ICD-10 | D66-D68 |
| ICD-9 | 286 |
| OMIM | 306700 306900 264900 |
| DiseasesDB | 5555 Template:DiseasesDB2 Template:DiseasesDB2 |
| MedlinePlus | 000537 |
| MeSH | D025861 |
|
WikiDoc Resources for Hemophilia |
|
Articles |
|---|
|
Most recent articles on Hemophilia |
|
Media |
|
Evidence Based Medicine |
|
Clinical Trials |
|
Ongoing Trials on Hemophilia at Clinical Trials.gov Clinical Trials on Hemophilia at Google
|
|
Guidelines / Policies / Govt |
|
US National Guidelines Clearinghouse on Hemophilia
|
|
Books |
|
News |
|
Commentary |
|
Definitions |
|
Patient Resources / Community |
|
Patient resources on Hemophilia Discussion groups on Hemophilia Patient Handouts on Hemophilia Directions to Hospitals Treating Hemophilia Risk calculators and risk factors for Hemophilia
|
|
Healthcare Provider Resources |
|
Causes & Risk Factors for Hemophilia |
|
Continuing Medical Education (CME) |
|
International |
|
|
|
Business |
|
Experimental / Informatics |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Please Take Over This Page and Apply to be Editor-In-Chief for this topic: There can be one or more than one Editor-In-Chief. You may also apply to be an Associate Editor-In-Chief of one of the subtopics below. Please mail us [2] to indicate your interest in serving either as an Editor-In-Chief of the entire topic or as an Associate Editor-In-Chief for a subtopic. Please be sure to attach your CV and or biographical sketch.
Overview
Hemophilia or haemophilia is the name of a family of hereditary genetic illnesses that impair the body's ability to control coagulation.
Differential Diagnosis
Hemophilia A can be mimicked by von Willebrand Disease
- von Willebrand Disease type 2A, where decreased levels of von Willebrand Factor can lead to premature proteolysis of Factor VIII. In contrast to haemophilia, vWD type 2A is inherited in an autosomal dominant fashion.
- von Willebrand Disease type 2N, where von Willebrand Factor cannot bind Factor VIII
- von Willebrand Disease type 3, where lack of von Willebrand Factor causes premature proteolysis of Factor VIII. In contrast to haemophilia, vWD type 3 is inherited in an autosomal recessive fashion.
Presentation
Genetic deficiencies and a rare autoimmune disorder may lower plasma clotting factor levels of coagulation factors needed for a normal clotting process. When a blood vessel is injured, a temporary scab does form, but the missing coagulation factors prevent fibrin formation which is necessary to maintain the blood clot. Therefore, there is no increase in bleeding time with haemophilia because platelets are intact, allowing the formation of these temporary hemostatic plugs (clots). However, "late" bleeding is affected, because these hemostatic plugs are not able to be maintained.
The bleeding with external injury is normal, but incidence of late re-bleeding and internal bleeding is increased, especially into muscles, joints, or bleeding into closed spaces. Major complications include hemarthrosis, hemorrhage, Gastrointestinal bleeding, and menorrhagia.
Causes
It is caused by a lack of clotting factors:
- Hemophilia A has a lack of the clotting Factor VIII. (Hemophilia A occurs in 90% of cases.)
- Hemophilia B has a lack of the clotting Factor IX.
- Hemophilia C has a lack of the clotting Factor XI.
History
The first record of haemophilia is in the Talmud, Jewish holy text, which states that males did not have to be circumcised if two brothers had already died from the procedure. In the 12th century, the Arab physician Albucasis wrote of a family whose males died of bleeding after minor injuries. Then, in 1803, Dr. John Conrad Otto, a Philadelphia physician, wrote an account about "a hemorrhagic disposition existing in certain families." He recognised that the disorder was hereditary and that it affected males and rarely females. He was able to trace the disease back to a woman who settled near Plymouth in 1720. The first usage of the term "haemophilia" appears in a description of the condition written by Hopff at the University of Zurich in 1828.[1] In 1937, Patek and Taylor, two doctors from Harvard, discovered anti-haemophilic globulin. Pavlosky, a doctor from Buenos Aires, found Hemophilia A and Hemophilia B to be separate diseases by doing a lab test. This test was done by transferring the blood of one haemophiliac to another haemophiliac. The fact that this corrected the clotting problem showed that there was more than one form of haemophilia.
- See main article at Haemophilia in European royalty
Haemophilia figured prominently in the history of European royalty and thus is sometimes known as "the royal disease". Queen Victoria passed the mutation to her son Leopold and, through several of her daughters, to various royals across the continent, including the royal families of Spain, Germany, and Russia. Tsarevich Alexei Nikolaevich, son of Nicholas II, was a descendant of Queen Victoria and suffered from haemophilia.
Prior to 1985, there were no laws enacted to screen blood, even though the technology existed. Corporations decided that the deaths of thousands of young men were more cost efficient than the instalation and usage of the screening equipment. As a result, many haemophilia patients who received untested and unscreened clotting factor prior to 1992 were at an extreme risk for contracting HIV and Hepatitis C via these blood products. At a rate of over 90% of the Haemophilia population, over 10,000 people contracted HIV from the tainted blood supply in the United States alone.
About 18,000 people in the United States have haemophilia. Each year, about 400 babies are born with the disorder. Haemophilia usually occurs in males and less often in females.
In the UK we now know that of every four baby boys born with Haemophilia, one will be born to a family with no previous history of the condition.
Genetic structure

Females possess two X-chromosomes, whereas males have one X and one Y chromosome. Since the mutations causing the disease are recessive, a woman carrying the defect on one of her X-chromosomes may not be affected by it, as the equivalent allele on her other chromosome should express itself to produce the necessary clotting factors. However the Y-chromosome in men has no gene for factors VIII or IX.
If the genes responsible for production of factor VIII or factor IX present on a male's X-chromosome is deficient there is no equivalent on the Y-chromosome, so the deficient gene is not masked by the dominant allele and he will develop the illness.
Since a male receives his single X-chromosome from his mother, the son of a healthy female silently carrying the deficient gene will have a 50% chance of inheriting that gene from her and with it the disease; and if his mother is affected with haemophilia, he will have a 100% chance of being a haemophiliac.
In contrast, for a female to inherit the disease, she must receive two deficient X-chromosomes, one from her mother and the other from her father (who must therefore be a haemophiliac himself). Hence haemophilia is far more common among males than females. However it is possible for female carriers to become mild haemophiliacs due to lyonisation of the X chromosomes.
Haemophiliac daughters are more common than they once were, as improved treatments for the disease have allowed more haemophiliac males to survive to adulthood and become parents. Adult females may experience menorrhagia (heavy periods) due to the bleeding tendency. The pattern of inheritance is criss-cross type. This type of pattern is also seen in color blindness.
As with all genetic disorders, it is of course also possible for a human to acquire it spontaneously (de novo), rather than inheriting it, because of a new mutation in one of their parents' gametes. Spontaneous mutations account for about ⅓ of all haemophilia A and 20% of all haemophilia B cases.
Genetic testing and genetic counseling is recommended for families with haemophilia. Prenatal testing, such as amniocentesis, is available to pregnant women who may be carriers of the condition.
Probability
If a female gives birth to a haemophiliac child, either the female is a carrier for the disease or the haemophilia was the result of a spontaneous mutation. Until modern direct DNA testing, however, it was impossible to determine if a female with only healthy children was a carrier or not. Generally, the more healthy sons she bore, the higher the probability that she was not a carrier. If the RH factor of the born male is different from the mother, the child will not be affected.
If a male is afflicted with the disease and has children, his daughters will be carriers of haemophilia. His sons, however, will not be affected with the disease. This is because the disease is X-linked and the father can not pass haemophilia through the Y chromosome.
Diagnostic Findings [2] [3]
Findings vary greatly with the different stages of hemophilic arthropathy (acute, subacute, or chronic hemarthrosis)
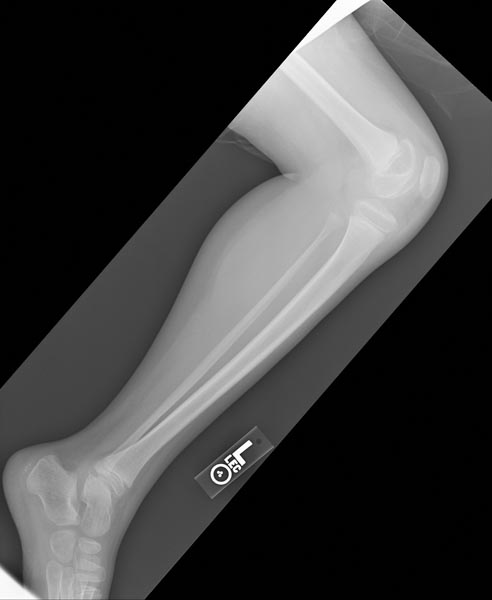
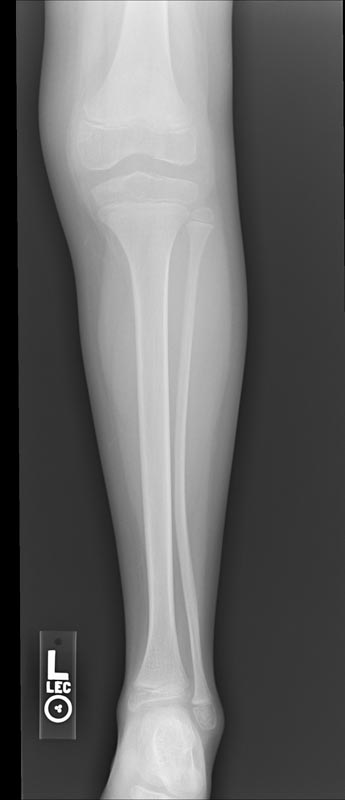
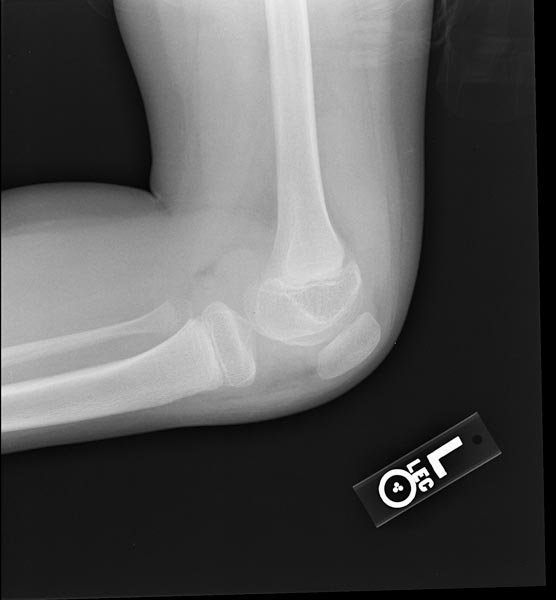
Images reflect the presence of hemarthrosis (joint effusion), synovial inflammation and hyperemia (osteoporosis and epiphyseal overgrowth), chondral erosions and subchondral resorption (osseous erosions and cysts), cartilaginous denudation (joint space narrowing), intraosseous or subperiosteal hemorrhage (pseudotumors), and osseous proliferation (sclerosis and osteophytes).
Some abnormalities of osseous shape, such as widening of the intercondylar notch, flattening of the condylar surface, or squaring of the patella, are very characteristic of chronic hemarthrosis of the knee.
- The clinical and radiologic features of patients with classic hemophilia and Christmas disease are virtually identical.
- Hemorrhage most often occurs in the synovial joints. In descending order, the knee, ankle, elbow, shoulder, and hip are involved.
- Repetitive bleeding into the musculoskeletal system is the most common complication of both conditions.
- Bleeding into the joints leads to hemophilic arthropathy.
- Bleeding into muscles causes joint contractures
- Bleeding into bone and adjacent soft tissues results in osseous and soft-tissue pseudotumors.
X-ray
-
Hemophilic arthropathy involving the bilateral knees
-
Hemophilic arthropathy involving the bilateral knees
-
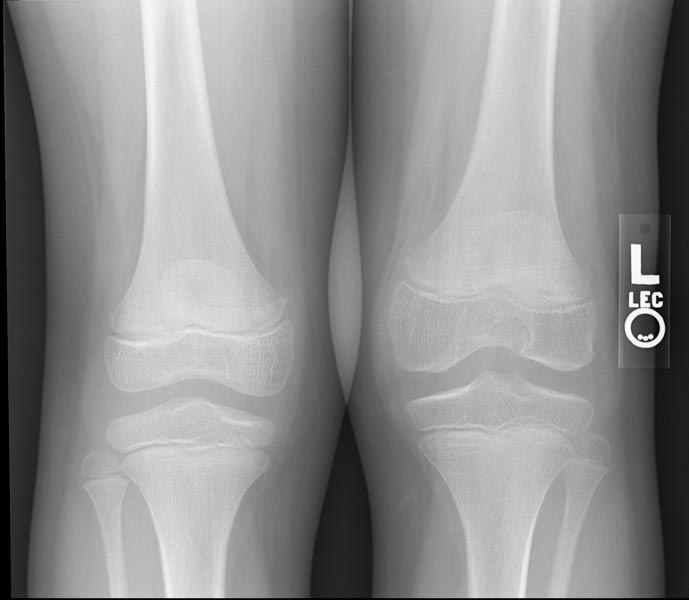
Hemophilic arthropathy involving the bilateral knees
-
Hemophilic arthropathy involving the bilateral knees




-
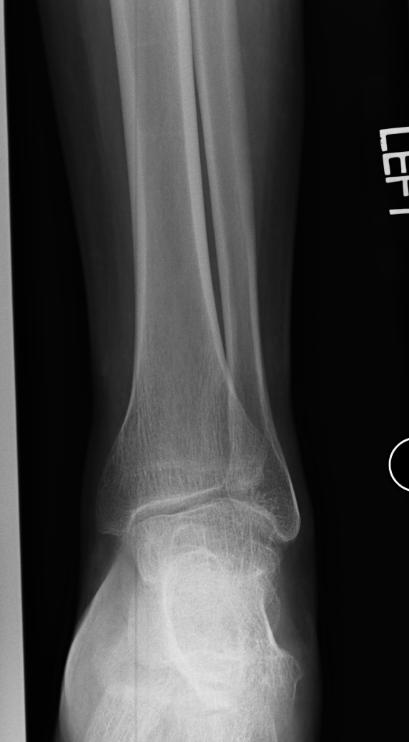
X-ray of the ankle in a patient with Hemophilia
-
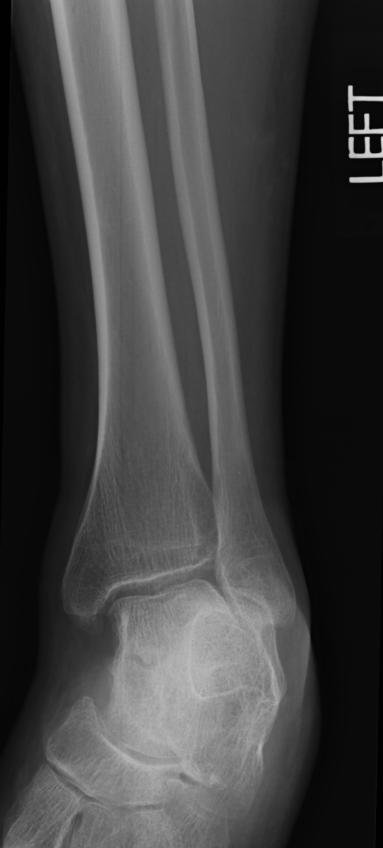
X-ray of the ankle in a patient with Hemophilia
-
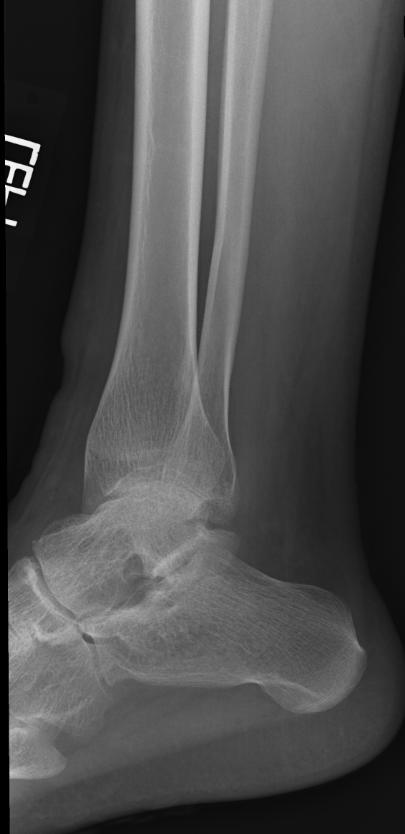
X-ray of the ankle in a patient with Hemophilia



-
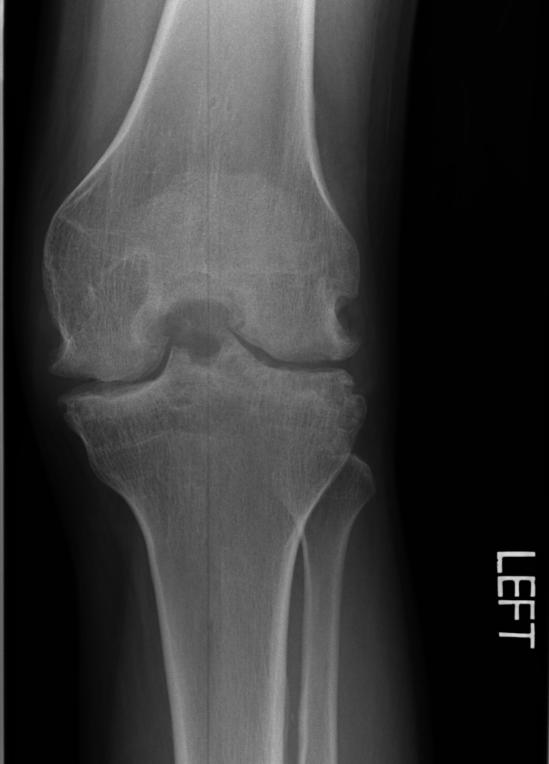
X-ray of the knee in a patient with Hemophilia
-
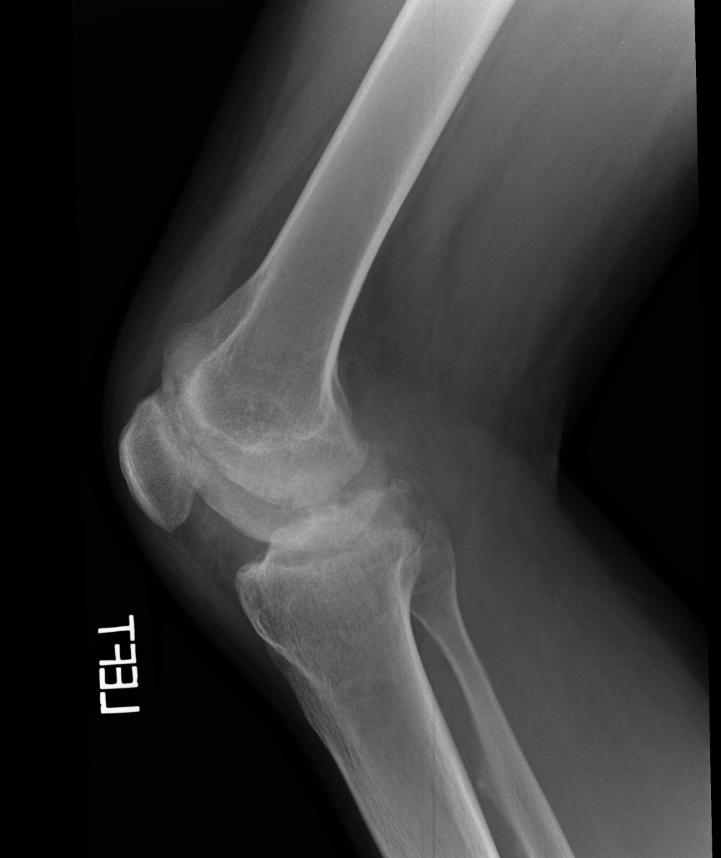
X-ray of the knee in a patient with Hemophilia


Computed Tomography
-
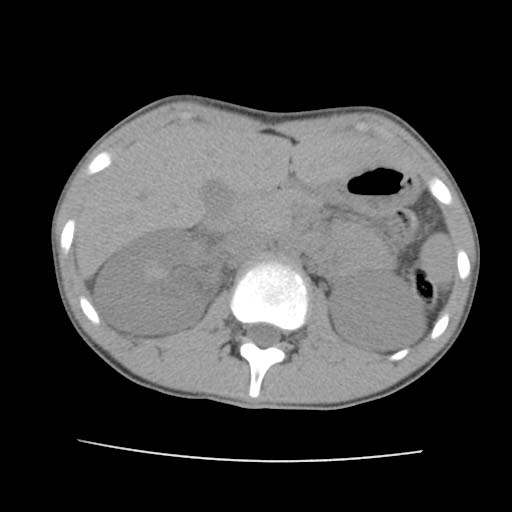
CT: Hemophilia. Bleeding into Kidney
-
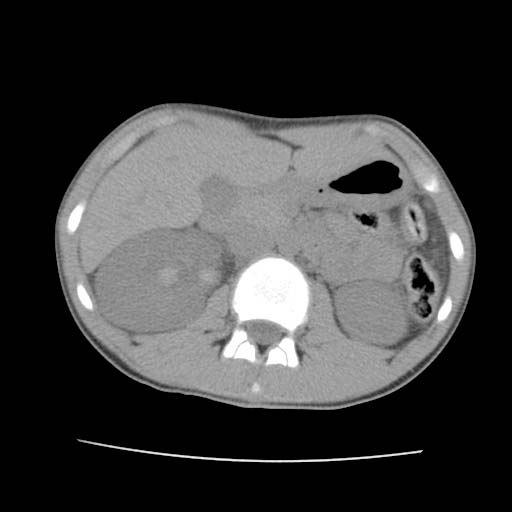
CT: Hemophilia. Bleeding into Kidney
-
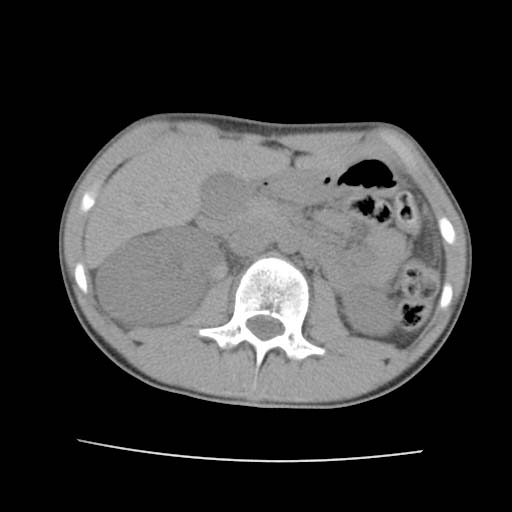
CT: Hemophilia. Bleeding into Kidney



MRI
-
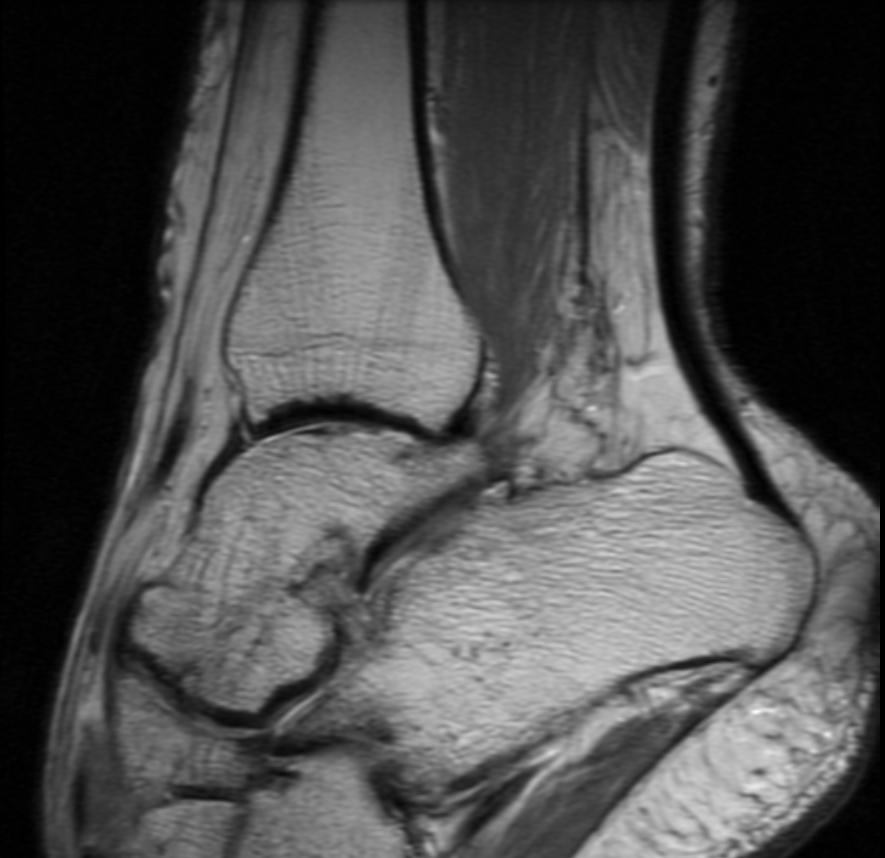
Ankle MRI in a patient with Hemophilia
-
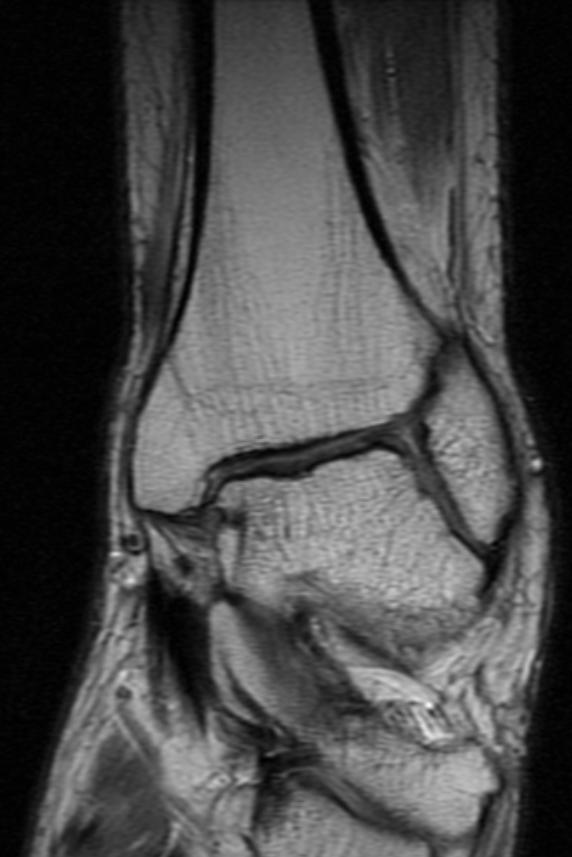
Ankle MRI in a patient with Hemophilia


-
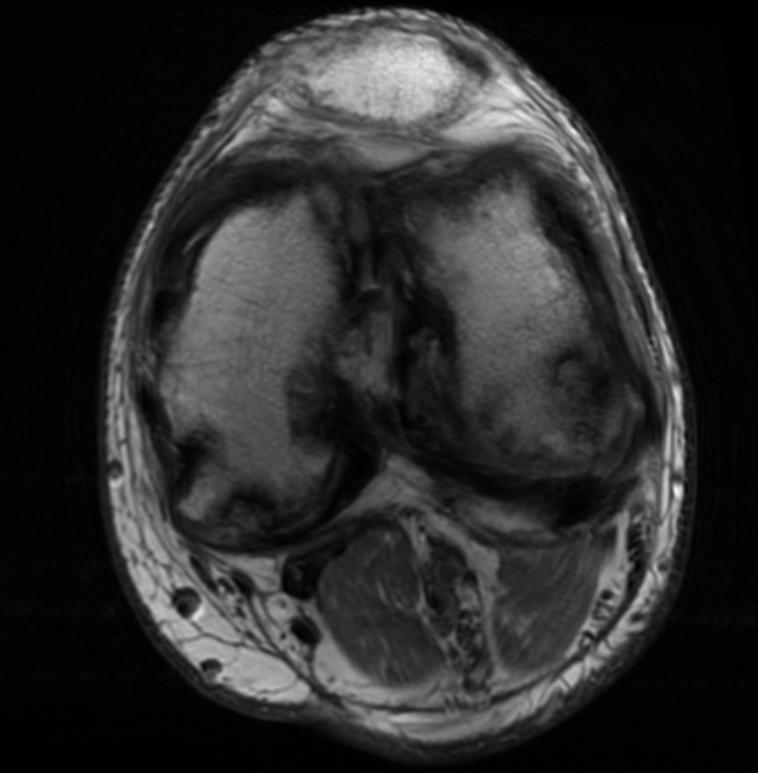
Knee MRI in a patient with Hemophilia
-
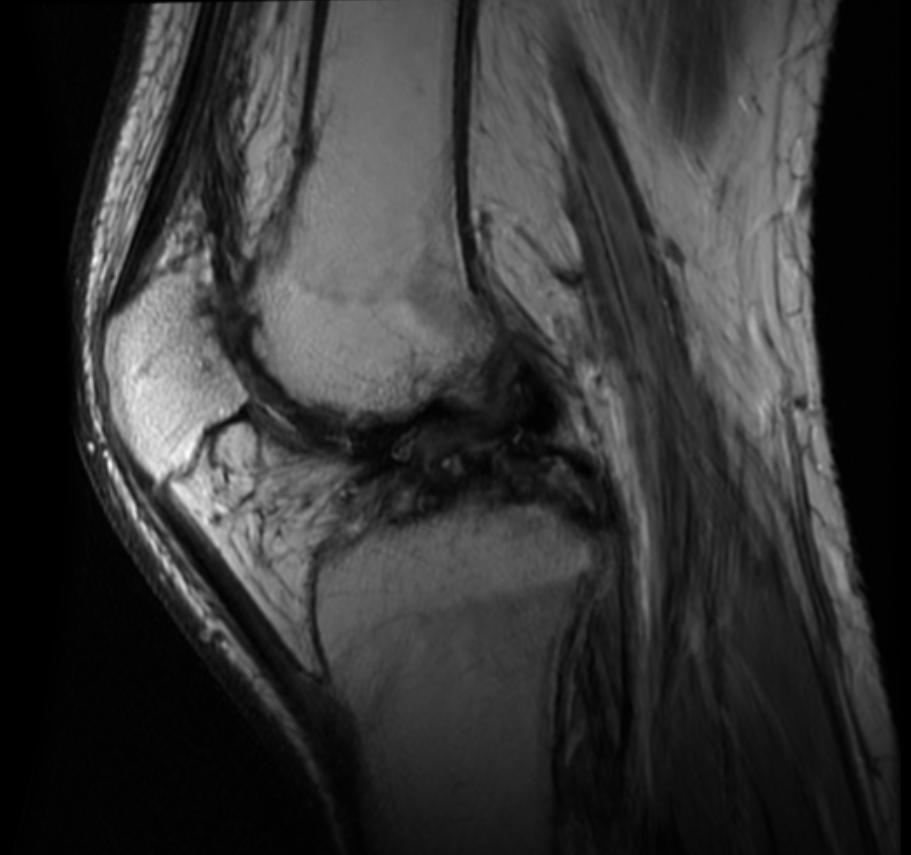
Knee MRI in a patient with Hemophilia
-
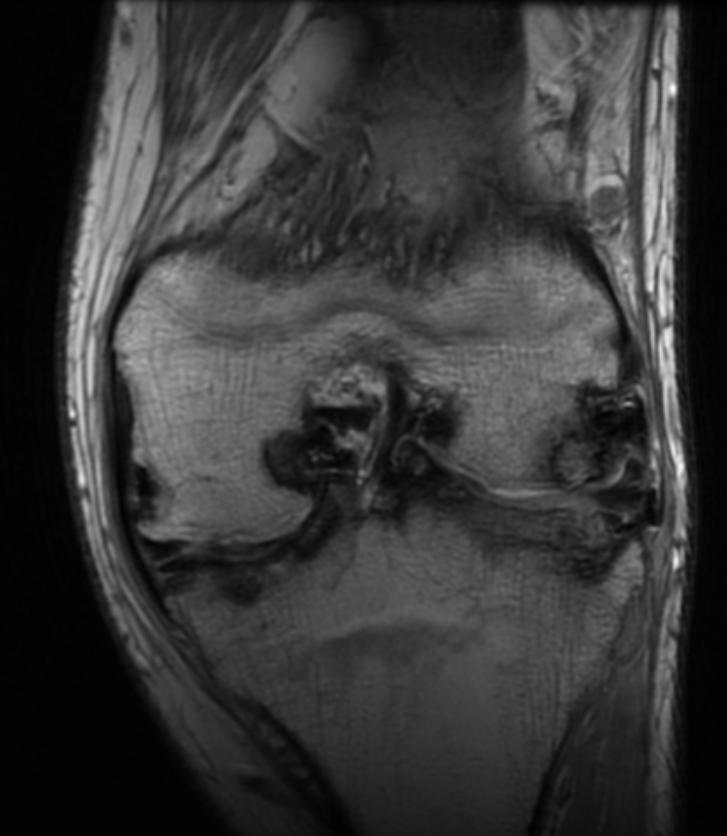
Knee MRI in a patient with Hemophilia



Treatment
Though there is no cure for haemophilia, it can be controlled with regular infusions of the deficient clotting factor, i.e. factor VIII in haemophilia A or factor IX in haemophilia B. Some haemophiliacs develop antibodies (inhibitors) against the replacement factors given to them, so the amount of the factor has to be increased or non-human replacement products must be given, such as porcine factor VIII Troy.
If a patient becomes refractory to replacement coagulation factor as a result of circulating inhibitors, this may be overcome with recombinant human factor VII (NovoSeven®), which is registered for this indication in many countries.
In western countries, common standards of care fall into one of two categories: prophylaxis or on-demand. Prophylaxis involves the infusion of clotting factor on a regular schedule in order to keep clotting levels sufficiently high to prevent spontaneous bleeding episodes. On-demand treatment involves treating bleeding episodes once they arise. In 2007, a clinical trial was published in the New England Journal of Medicine comparing on-demand treatment of boys (< 30 months) with Haemophilia A with prophylactic treatment (infusions of 25 IU/kg body weight of Factor VIII every other day) in respect to its effect on the prevention of joint-diseases. When the boys reached 6 years of age, 93% of those in the prophylaxis group and 55% of those in the episodic-therapy group had a normal index joint-structure on MRI. [4] Prophylactic treatment, however, resulted in average costs of $300,000 per year. The author of an editorial published in the same issue of the New England Journal of Medicine demands more clinical studies addressing the cost-effectiveness of prophylactic treatment. [5]
As a direct result of the contamination of the blood supply in the late 1970s and early/mid 1980s with viruses such as Hepatitis and HIV, new methods were developed in the production of clotting factor products. The initial response was to heat-treat (pasteurize) plasma-derived factor concentrate, followed by the development of monoclonal factor concentrates, which use a combination of heat treatment and affinity chromatography to inactivate any viral agents in the pooled plasma from which the factor concentrate is derived. The Lindsay Tribunal in Ireland investigated, among other things, the slow adoption of the new methods.
Since 1993 (Dr. Mary Nugent), recombinant factor products (which are typically cultured in Chinese hamster ovary (CHO) tissue culture cells and involve little, if any human plasma products) have become available and are widely used in wealthier western countries. While recombinant clotting factor products offer higher purity and safety, they are, like concentrate, extremely expensive, and not generally available in the developing world. In many cases, factor products of any sort are difficult to obtain in developing countries.
It was claimed that Rasputin was successful at treating the Tsarevich Alexei of Russia's haemophilia: however, to this day it is unclear how he accomplished this.
References
- ↑ "The History of haemophilia". Retrieved 2007-06-27.
- ↑ Hermann, G, Gilbert, MS, Abdelwahab, IF Hemophilia: evaluation of musculoskeletal involvement with CT, sonography, and MR imaging. Am. J. Roentgenol. 1992 158: 119-123.
- ↑ Jaume Llauger, Jaume Palmer, Núria Rosón, Sílvia Bagué, Àngels Camins, and Rosa Cremades. Nonseptic Monoarthritis: Imaging Features with Clinical and Histopathologic Correlation. RadioGraphics 2000 20: 263S-278S.
- ↑ Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe haemophilia. N Engl J Med. 2007 Aug 9;357(6):535-44. PMID 17687129
- ↑ Roosendaal G, Lafeber F. Prophylactic treatment for prevention of joint disease in haemophilia--cost versus benefit. N Engl J Med. 2007 Aug 9;357(6):603-5. PMID 17687136
Template:Hematology Template:SIB
af:Hemofilie
ar:نزف الدم الوراثي
ca:Hemofília
cs:Hemofilie
de:Hämophilie
eo:Hemofilio
ko:혈우병
hr:Hemofilija
id:Hemofilia
ia:Hemophilia
it:Emofilia
he:המופיליה
nl:Hemofilie
no:Hemofili
sr:Хемофилија
fi:Verenvuototauti
sv:Blödarsjuka
ta:இரத்தம் உறையாமை
ur:انس الدم