Myelofibrosis: Difference between revisions
No edit summary |
No edit summary |
||
| Line 22: | Line 22: | ||
'''Myelofibrosis with myeloid metaplasia''', also known as '''agnogenic myeloid metaplasia''', '''chronic idiopathic myelofibrosis''', and '''primary myelofibrosis''',<ref>Older terms include "myelofibrosis with myeloid metaplasia" and "agnogenic myeloid metaplasia". The [[World Health Organization]] utilizes the name "chronic idiopathic myelofibrosis", while the International Working Group on Myelofibrosis Research and Treatment calls the disease "primary myelofibrosis".</ref> was first described in 1879 and is currently classified as a [[myeloproliferative disease]] caused by the growth and proliferation of an abnormal [[bone marrow]] stem cell, resulting in the [[fibrosis|replacement of the bone marrow with fibrous connective tissue]]. An eponym for the disease is Heuck-Assmann disease, or Assmann's Disease. | '''Myelofibrosis with myeloid metaplasia''', also known as '''agnogenic myeloid metaplasia''', '''chronic idiopathic myelofibrosis''', and '''primary myelofibrosis''',<ref>Older terms include "myelofibrosis with myeloid metaplasia" and "agnogenic myeloid metaplasia". The [[World Health Organization]] utilizes the name "chronic idiopathic myelofibrosis", while the International Working Group on Myelofibrosis Research and Treatment calls the disease "primary myelofibrosis".</ref> was first described in 1879 and is currently classified as a [[myeloproliferative disease]] caused by the growth and proliferation of an abnormal [[bone marrow]] stem cell, resulting in the [[fibrosis|replacement of the bone marrow with fibrous connective tissue]]. An eponym for the disease is Heuck-Assmann disease, or Assmann's Disease. | ||
== | ==Pathophysiology== | ||
== Signs and symptoms == | The bone marrow is replaced by collagen fibrosis, impairing the patient's ability to generate new blood cells resulting in a progressive [[pancytopenia]]. It is usually reactive following other [[myeloproliferative disorders]], such as [[polycythemia rubra vera]] or [[essential thrombocytosis]]. [[Extramedullary haematopoeisis]] occurs as the haemopoetic cells migrate away from the bone marrow, to the liver and spleen. Patients often have [[hepatosplenomegaly]] and [[poikilocytosis]]. | ||
In primary myelofibrosis, a progressive scarring ([[fibrosis]]) of the bone marrow occurs. As a result, blood forms in sites other than the bone marrow, such as the [[liver]] and [[spleen]]. This causes an enlargement of these organs. The cause and risk factors are unknown. It commonly occurs in the spent phase of [[Polycythemia rubra vera]], possibly in response to the medication [[hydroxyurea]] poisoning the marrow. | |||
Genetic associations with [[JAK2]]<ref name="pmid15781101">{{cite journal |author=Baxter EJ, Scott LM, Campbell PJ, ''et al'' |title=Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders |journal=Lancet |volume=365 |issue=9464 |pages=1054–61 |year=2005 |pmid=15781101 |doi=10.1016/S0140-6736(05)71142-9 |url=http://linkinghub.elsevier.com/retrieve/pii/S0140-6736(05)71142-9}}</ref> and [[Myeloproliferative leukemia virus oncogene|MPL]]<ref name="pmid16834459">{{cite journal |author=Pikman Y, Lee BH, Mercher T, ''et al'' |title=MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia |journal=PLoS Med. |volume=3 |issue=7 |pages=e270 |year=2006 |month=July |pmid=16834459 |pmc=1502153 |doi=10.1371/journal.pmed.0030270 |url=http://medicine.plosjournals.org/perlserv/?request=get-document&doi=10.1371/journal.pmed.0030270}}</ref> have been described. | |||
== Signs and symptoms == | |||
*Abdominal fullness related to an [[splenomegaly|enlarged spleen]] (splenomegaly). | *Abdominal fullness related to an [[splenomegaly|enlarged spleen]] (splenomegaly). | ||
*Bone pain | *Bone pain | ||
Revision as of 14:18, 19 January 2009
Template:DiseaseDisorder infobox Template:Search infobox Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Please Take Over This Page and Apply to be Editor-In-Chief for this topic: There can be one or more than one Editor-In-Chief. You may also apply to be an Associate Editor-In-Chief of one of the subtopics below. Please mail us [2] to indicate your interest in serving either as an Editor-In-Chief of the entire topic or as an Associate Editor-In-Chief for a subtopic. Please be sure to attach your CV and or biographical sketch.
Overview
Myelofibrosis with myeloid metaplasia, also known as agnogenic myeloid metaplasia, chronic idiopathic myelofibrosis, and primary myelofibrosis,[1] was first described in 1879 and is currently classified as a myeloproliferative disease caused by the growth and proliferation of an abnormal bone marrow stem cell, resulting in the replacement of the bone marrow with fibrous connective tissue. An eponym for the disease is Heuck-Assmann disease, or Assmann's Disease.
Pathophysiology
The bone marrow is replaced by collagen fibrosis, impairing the patient's ability to generate new blood cells resulting in a progressive pancytopenia. It is usually reactive following other myeloproliferative disorders, such as polycythemia rubra vera or essential thrombocytosis. Extramedullary haematopoeisis occurs as the haemopoetic cells migrate away from the bone marrow, to the liver and spleen. Patients often have hepatosplenomegaly and poikilocytosis.
In primary myelofibrosis, a progressive scarring (fibrosis) of the bone marrow occurs. As a result, blood forms in sites other than the bone marrow, such as the liver and spleen. This causes an enlargement of these organs. The cause and risk factors are unknown. It commonly occurs in the spent phase of Polycythemia rubra vera, possibly in response to the medication hydroxyurea poisoning the marrow.
Genetic associations with JAK2[2] and MPL[3] have been described.
Signs and symptoms
- Abdominal fullness related to an enlarged spleen (splenomegaly).
- Bone pain
- Bruising and easy bleeding due to inadequate numbers of platelets
- Fatigue
- Increased susceptibility to infection, such as pneumonia or diarrhea
- Pallor and shortness of breath while doing physical work due to anemia
Diagnosis
Diagnosis is based upon:
- Normochromic normocytic anaemia
- Red cell poikilocytosis on blood film (tear drop RBCs)
- JAK 2 mutation on Val 617 Phe locus in 50%
- Raised lactate dehydrogenase
- Raised neutrophil alkaline phosphatase score
- Bone marrow biopsy may show increased cellularity and fibrosis
Diagnostic Findings
MRI
-

Myelofibrosis
-
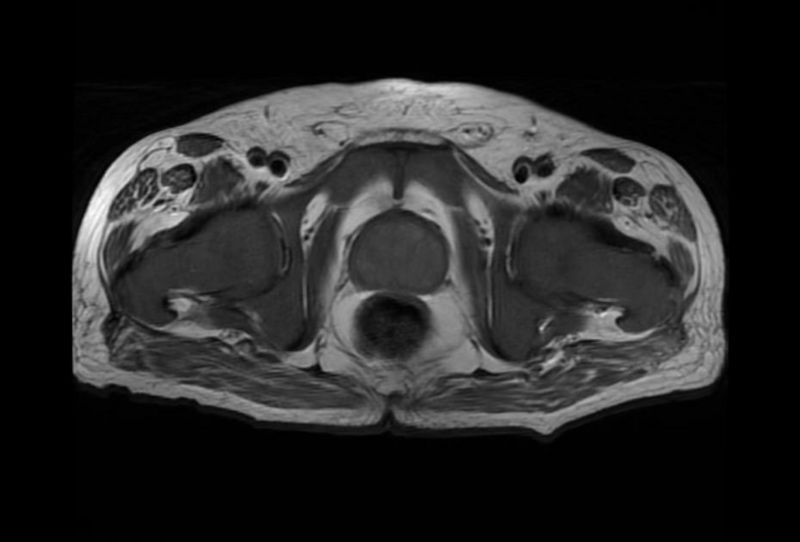
Myelofibrosis


-

Myelofibrosis
-
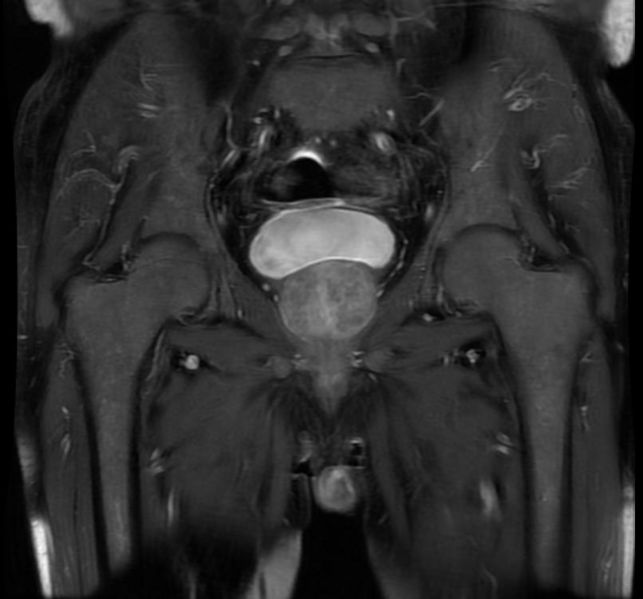
Myelofibrosis


References
- ↑ Older terms include "myelofibrosis with myeloid metaplasia" and "agnogenic myeloid metaplasia". The World Health Organization utilizes the name "chronic idiopathic myelofibrosis", while the International Working Group on Myelofibrosis Research and Treatment calls the disease "primary myelofibrosis".
- ↑ Baxter EJ, Scott LM, Campbell PJ; et al. (2005). "Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders". Lancet. 365 (9464): 1054–61. doi:10.1016/S0140-6736(05)71142-9. PMID 15781101.
- ↑ Pikman Y, Lee BH, Mercher T; et al. (2006). "MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia". PLoS Med. 3 (7): e270. doi:10.1371/journal.pmed.0030270. PMC 1502153. PMID 16834459. Unknown parameter
|month=ignored (help)