Cerebral hypoxia pathophysiology
|
Cerebral hypoxia Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Cerebral hypoxia pathophysiology On the Web |
|
American Roentgen Ray Society Images of Cerebral hypoxia pathophysiology |
|
Risk calculators and risk factors for Cerebral hypoxia pathophysiology |
Overview
The brain consumes significant amount of the energy compared to its size and weight. Cellular injury can begin within minutes, and permanent brain injury will follow if prompt intervention does not occur.
Pathophysiology
- The brain depends on a constant energy supply provided by glucose and oxygen but is unable to store energy. With the cessation of blood flow, intracellular production of adenosine triphosphate is diminished. This results in dysfunction of energy-dependent ion channels, which contributes to intracellular sodium accumulation and cytotoxic edema. Ongoing ischemia results in the release of glutamate, an excitatory neurotransmitter, which promotes calcium influx through N-methyl-D-aspartate (NMDA) receptors. Calcium influx exacerbates neuronal injury by activating lytic enzymes, precipitating free radical formation, and interfering with mitochondrial function. This process, known as excitotoxicity, can ultimately lead to cell death. [6][7]
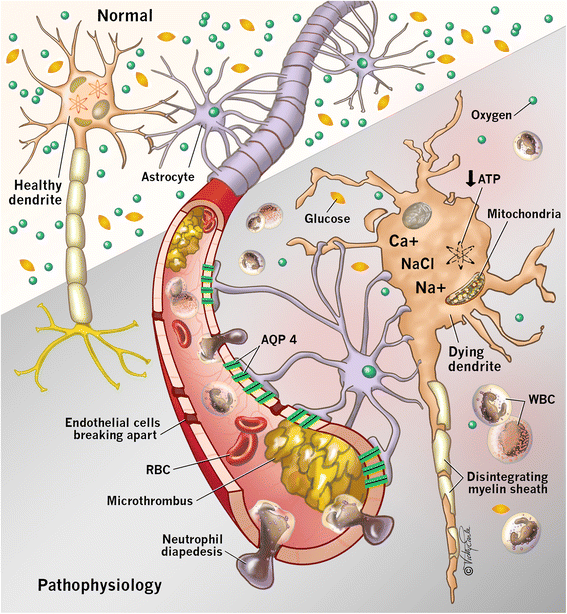
Pathophysiology
