|
|
| Line 1: |
Line 1: |
| <div style="-webkit-user-select: none;"> | | <div style="-webkit-user-select: none;"> |
| {|class="infobox" style="position: fixed; top: 65%; right: 10px; margin: 0 0 0 0; border: 0; float: right; | | {| class="infobox" style="position: fixed; top: 65%; right: 10px; margin: 0 0 0 0; border: 0; float: right;" |
| |- | | |- |
| | {{#ev:youtube|https://https://www.youtube.com/watch?v=v5gdH_Hydes|350}} | | | {{#ev:youtube|https://https://www.youtube.com/watch?v=v5gdH_Hydes|350}} |
| Line 9: |
Line 9: |
|
| |
|
| ==Overview== | | ==Overview== |
|
| |
| Alzheimer's disease develops from the loss of [[neurons]] and [[synapses|synaptic connections]]. There exist at least three major hypotheses that aim to describe the mechanism of how Alzheimer's disease proliferates in the [[brain]].
| |
|
| |
|
| ==Pathogenesis== | | ==Pathogenesis== |
| Line 16: |
Line 14: |
| [[Image:Alzheimer's disease - MRI.jpg|left|150x200px|frame|[[MRI]] images of a normal aged brain (right) and an Alzheimer's patient's brain (left). In the Alzheimer brain, atrophy is clearly seen.]] | | [[Image:Alzheimer's disease - MRI.jpg|left|150x200px|frame|[[MRI]] images of a normal aged brain (right) and an Alzheimer's patient's brain (left). In the Alzheimer brain, atrophy is clearly seen.]] |
|
| |
|
| At a [[macroscopic]] level, AD is characterized by loss of [[neuron]]s and [[synapse]]s in the [[cerebral cortex]] and certain subcortical regions. This results in gross [[atrophy]] of the affected regions, including [[degeneration]] in the [[temporal lobe]] and [[parietal lobe]], and parts of the [[frontal cortex]] and [[cingulate gyrus]].<ref name="pmid12934968">{{cite journal |author=Wenk GL |title=Neuropathologic changes in Alzheimer's disease |journal=Journal of Clinical Psychiatry |volume=64 Suppl 9 |issue= |pages=7–10 |year=2003 |pmid=12934968 |doi=}}</ref>
| |
| Three major hypotheses exist to explain the cause of the disease, though other possible explanations also exist.
| |
|
| |
| ===Cholinergic hypothesis===
| |
|
| |
| The oldest major hypothesis is the ''[[cholinergic]] hypothesis'', which proposes that AD is caused by reduced synthesis of the [[neurotransmitter]] [[acetylcholine]]. Most currently available drug therapies in Alzheimer's are based on this theory, although the medications that treat [[acetylcholine]] deficiency only affect symptoms of the disease and will neither halt nor reverse the progression of AD.<ref name="pmid16644763">{{cite journal |author=Walker LC, Rosen RF |title=Alzheimer therapeutics-what after the cholinesterase inhibitors? |journal=Age Ageing |volume=35 |issue=4 |pages=332–335 |year=2006 |pmid=16644763 |doi=10.1093/ageing/afl009}}</ref>
| |
| The [[cholinergic]] hypothesis has not maintained widespread support in the face of this evidence, although cholinergic effects have been proposed to initiate large-scale aggregation,<ref name="pmid15236795">{{cite journal |author=Shen ZX |title=Brain cholinesterases: II. The molecular and cellular basis of Alzheimer's disease |journal=Medical Hypotheses |volume=63 |issue=2 |pages=308–321 |year=2004 |pmid=15236795 |doi=10.1016/j.mehy.2004.02.031}}</ref> leading to generalized neuroinflammation.<ref name="pmid12934968">{{cite journal |author=Wenk GL |title=Neuropathologic changes in Alzheimer's disease |journal=Journal of Clinical Psychiatry |volume=64 Suppl 9 |issue= |pages=7–10 |year=2003 |pmid=12934968 |doi=}}</ref> In 1991 the [[amyloid beta|amyloid]] hypothesis was proposed,<ref name="pmid1763432">{{cite journal |author=Hardy J, Allsop D |title=Amyloid deposition as the central event in the aetiology of Alzheimer's disease |journal=Trends Pharmacol. Sci. |volume=12 |issue=10 |pages=383–8 |year=1991 |pmid=1763432 |doi=10.1016/0165-6147(91)90609-V }}</ref> while research after 2000 is also centered on [[tau protein]]s. The two positions differ insofar as one states that the [[tau protein]] abnormalities initiate the disease cascade, while the other states that [[amyloid beta]] (Aβ) deposits are the causative factor in the disease.<ref name="pmid11801334">{{cite journal |author=Mudher A, Lovestone S |title=Alzheimer's disease-do tauists and baptists finally shake hands? |journal=Trends in Neuroscience |volume=25 |issue=1 |pages=22–26 |year=2002 |pmid=11801334 | doi=10.1016/S0166-2236(00)02031-2 }}</ref> Other changes, including congophilic amyloid angiopathy, oxidative changes, neuronal loss, and [[inflammation]] are also associated with Alzheimer's disease.
| |
|
| |
| ===Amyloid hypothesis===
| |
|
| |
| [[Image:Amyloid-plaque formation-big.jpg|left|50x100px|frame|Enzymes act on the APP (amyloid precursor protein) and cut it into fragments. The beta-amyloid fragment is crucial in the formation of senile plaques in AD.]]
| |
|
| |
| Alzheimer's disease has been identified as a [[protein folding|protein misfolding]] disease, or [[proteopathy]], due to the accumulation of abnormally folded A-beta and tau proteins in the brains of AD patients.<ref name="pmid14528050">{{cite journal |author=Hashimoto M, Rockenstein E, Crews L, Masliah E |title=Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases |journal=Neuromolecular Medicine |volume=4 |issue=1–2 |pages=21–36 |year=2003 |pmid=14528050 |doi=10.1385/NMM:4:1-2:21}}</ref> The ''[[amyloid beta|amyloid]] hypothesis'' postulates that amyloid beta (Aβ) deposits are the fundamental cause of the disease.<ref name="pmid1763432">{{cite journal
| |
| |author=Hardy J, Allsop D
| |
| |title=Amyloid deposition as the central event in the aetiology of Alzheimer's disease
| |
| |journal=Trends Pharmacol. Sci.
| |
| |volume=12
| |
| |issue=10
| |
| |pages=383–88
| |
| |year=1991
| |
| |month=October
| |
| |pmid=1763432
| |
| }}</ref><ref name="pmid11801334">{{cite journal
| |
| |author=Mudher A, Lovestone S
| |
| |title=Alzheimer's disease-do tauists and baptists finally shake hands?
| |
| |journal=Trends Neurosci.
| |
| |volume=25
| |
| |issue=1
| |
| |pages=22–26
| |
| |year=2002
| |
| |month=January
| |
| |pmid=11801334
| |
| }}</ref>
| |
| [[Plaques]] are made of a small [[peptide]] (39 to 43 amino acid residues) called [[beta-amyloid]] (also A-beta or Aβ), a [[protein]] fragment snipped from a larger protein called [[amyloid precursor protein]] (APP). APP is a [[transmembrane protein]]; which means that it sticks through the neuron's membrane; and is believed to help neurons grow, survive and repair themselves after injury.<ref name="pmid16822978">{{cite journal |author=Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J |title=Synapse formation and function is modulated by the amyloid precursor protein |journal=Journal of Neuroscience |volume=26 |issue=27 |pages=7212–7221 |year=2006 |pmid=16822978 |doi=10.1523/JNEUROSCI.1450-06.2006}}</ref><ref name="pmid12927332">{{cite journal |author=Turner PR, O'Connor K, Tate WP, Abraham WC |title=Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory |journal=Prog. Neurobiology |volume=70 |issue=1 |pages=1–32 |year=2003 |pmid=12927332 |doi=}}</ref> In AD, APP is divided by [[enzymes]] such as [[gamma-secretase]] and [[BACE1]] through a mechanism called [[proteolysis]].<ref name="pmid15787600">{{cite journal |author=Hooper NM |title=Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein |journal=Biochemical Society Transactions |volume=33 |issue=Pt 2 |pages=335–338 |year=2005 |pmid=15787600 |doi=10.1042/BST0330335}}</ref> One of these fragments is [[beta-amyloid]]. Beta-amyloid fragments (amyloid fibrils) outside the cell form clumps that deposit outside neurons in dense formations known as [[senile plaques]].<ref name="pmid15004691">{{cite journal |author=Ohnishi S, Takano K |title=Amyloid fibrils from the viewpoint of protein folding |journal=Cellular Molecular Life Sciences |volume=61 |issue=5 |pages=511–524 |year=2004 |pmid=15004691 |doi=10.1007/s00018-003-3264-8}}</ref><ref name="pmid15184601">{{cite journal |author=Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J |title=The importance of neuritic plaques and tangles to the development and evolution of AD |journal=Neurology |volume=62 |issue=11 |pages=1984–1989 |year=2004 |pmid=15184601 |doi=}}</ref>
| |
| The amyloid hypothesis is compelling because the gene for the amyloid beta precursor (APP) is located on [[chromosome 21]], and patients with [[trisomy 21]] ([[Down Syndrome]]) who thus have an extra [[gene dosage|gene copy]] almost universally exhibit AD-like disorders by 40 years of age.<ref name="pmid16904243">{{cite journal
| |
| |author=Nistor M, Don M, Parekh M, Sarsoza F, Goodus M, Lopez GE, Kawas C, Leverenz J, Doran E, Lott IT, Hill M, Head E
| |
| |title=Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain
| |
| |journal=Neurobiol. Aging
| |
| |volume=28
| |
| |issue=10
| |
| |pages=1493–506
| |
| |year=2007
| |
| |pmid=16904243
| |
| |doi=10.1016/j.neurobiolaging.2006.06.023
| |
| }}</ref><ref name="pmid15639317">{{cite journal |author=Lott IT, Head E |title=Alzheimer disease and Down syndrome: factors in pathogenesis |journal=Neurobiology of Aging |volume=26 |issue=3 |pages=383–389 |year=2005 |pmid=15639317 |doi=10.1016/j.neurobiolaging.2004.08.005}}</ref> It should be noted further that [[ApoE4]], the major genetic risk factor for AD, leads to excess amyloid build-up in the brain before AD symptoms arise. Thus, Aβ deposition precedes clinical AD.<ref name="pmid7566000">{{cite journal
| |
| |author=Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, Niinistö L, Halonen P, Kontula K
| |
| |title=Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein
| |
| |journal=New England Journal of Medicine
| |
| |volume=333
| |
| |issue=19
| |
| |pages=1242–1247
| |
| |year=1995
| |
| |pmid=7566000
| |
| |doi=10.1056/NEJM199511093331902
| |
| }}</ref> It is known that some types of inherited AD involve only mutations in the APP gene (although this is not the most common type—others involve genes for "pre-senilin" proteins which process APP and may also have still-unknown functions).<ref>{{cite web |url=http://ghr.nlm.nih.gov/condition=alzheimerdisease |title=Alzheimer disease |publisher=US National Library of Medicine |date=2008-04-25 |accessdate=2008-05-02}}</ref> However, another strong support for the amyloid hypothesis, which looks at Aβ as the common initiating factor for Alzheimer's disease, is that [[Genetically modified organism|transgenic]] mice solely expressing a mutant human APP gene develop fibrillar [[amyloid plaques]].<ref>Beta-amyloid precursor protein
| |
| * {{cite journal
| |
| |author=Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F
| |
| |title=Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein
| |
| |journal=Nature
| |
| |volume=373
| |
| |issue=6514
| |
| |pages=523–527
| |
| |year=1995
| |
| |pmid=7845465
| |
| |doi=10.1038/373523a0
| |
| }}
| |
| * {{cite journal |author=Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D |title=Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease |journal=Journal of Neuroscience |volume=16 |issue=18 |pages=5795–5811 |year=1996 |pmid=8795633 |doi=}}
| |
| * {{cite journal
| |
| |author=Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G
| |
| |title=Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice
| |
| |journal=Science
| |
| |volume=274
| |
| |issue=5284
| |
| |pages=99–102
| |
| |year=1996
| |
| |pmid=8810256
| |
| |doi = 10.1126/science.274.5284.99
| |
| }}</ref>
| |
| If damage from [[Aβ]] is the primary initiating cause of AD, the exact mechanism has not been elucidated. The traditional formulation of the amyloid hypothesis points to the [[cytotoxicity]] of mature aggregated amyloid fibrils.<ref name="pmid2218531">{{cite journal |author=Yankner BA, Duffy LK, Kirschner DA |title=Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides |journal=Science |volume=250 |issue=4978 |pages=279–282 |year=1990 |pmid=2218531 |doi=10.1126/science.2218531 }}</ref> The most [[neurotoxic]] form of [[amyloid]] are the soluble [[oligomers]] and intermediate amyloids; the severity of cognitive defect in AD correlates with oligomeric level in [[brain tissue]]. It is also known that [[Aβ]] selectively builds up in the [[mitochondria]] of samples from the [[brains]] of humans with AD, as well as in [[mitochondria]] from transgenic mice with APP genes. In both cases, it inhibits certain mitochondrial enzyme functions and exhibits a similar decrease in glucose utilization in neurons to the one which is a known characteristic of AD. This process may also lead to the formation of damaging [[reactive oxygen species]], [[calcium]] influx, and [[apoptosis]]. Mechanisms which involve direct damage from [[Aβ]] before it forms fibrils and [[plaques]] also address the issue that [[neuronal]] damage is not correlated as well with [[plaques]], since in this model it is not the plaques themselves which cause the major damage, but rather the precursor [[Aβ]] protein directly, via another mechanism.<ref name="pmid17424907">{{cite journal|author=Chen, X, Yan, SD|title=Mitochondrial Aβ: A Potential Cause of Metabolic Dysfunction in Alzheimer's Disease. |journal=IUBMB Life|volume=58|issue=12|pages=686-694|year=2006|pmid=17424907|doi=10.1080/15216540601047767}}</ref>
| |
| Again, deposition of amyloid plaques does not correlate well with neuron loss.<ref name="pmid15039236">{{cite journal
| |
| |author=Schmitz C, Rutten BP, Pielen A, ''et al''
| |
| |title=Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease
| |
| |journal=Am. J. Pathol.
| |
| |volume=164
| |
| |issue=4
| |
| |pages=1495–1502
| |
| |year=2004
| |
| |month=April
| |
| |pmid=15039236
| |
| |pmc=1615337
| |
| }}</ref>
| |
|
| |
| ===Tau hypothesis===
| |
| <gallery> | | <gallery> |
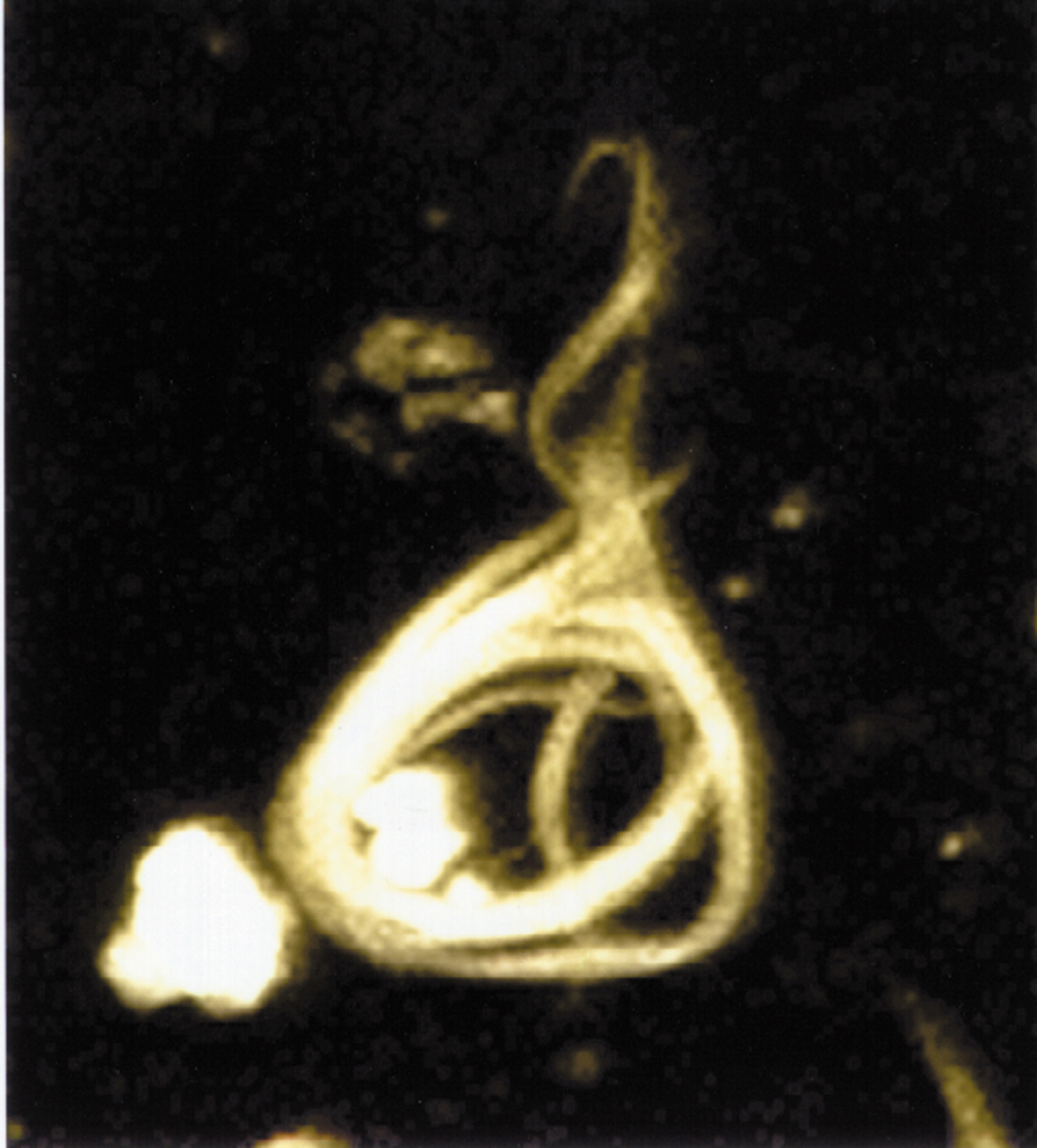
| Image:TAU HIGH.JPG|Microscopy image of a neurofibrillary tangle, conformed by hyperphosphorylated tau protein. | | Image:TAU HIGH.JPG|Microscopy image of a neurofibrillary tangle, conformed by hyperphosphorylated tau protein. |
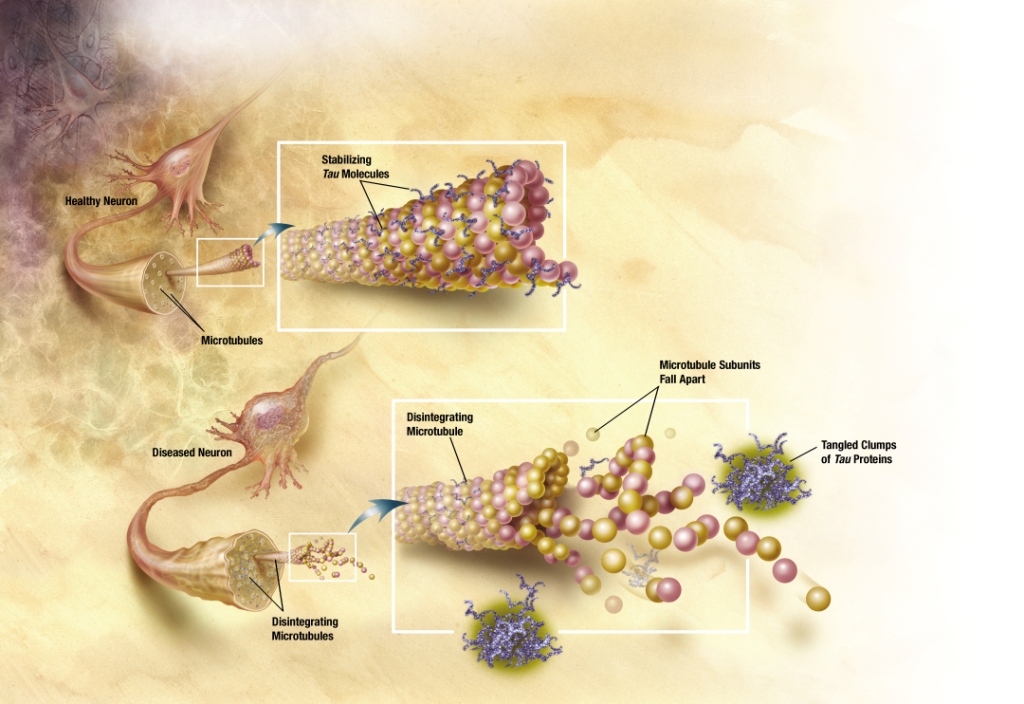
| Image:TANGLES HIGH.jpg|In Alzheimer's disease, changes in tau protein lead to the disintegration of microtubules in brain cells. | | Image:TANGLES HIGH.jpg|In Alzheimer's disease, changes in tau protein lead to the disintegration of microtubules in brain cells. |
| </gallery> | | </gallery> |
| The observation that [[amyloid plaques]] do not correlate with neuron loss supports the ''tau hypothesis''. The ''tau hypothesis'' supports the idea that [[tau protein]] abnormalities initiate the disease cascade.<ref name="pmid11801334"/> AD is also considered a [[tauopathy]] due to abnormal aggregation of the [[tau protein]]. Healthy neurons have an internal support structure, or [[cytoskeleton]], partly made up of structures called [[microtubules]]. These microtubules act like tracks, guiding nutrients and molecules from the body of the cell down to the ends of the [[axon]] and back. A special kind of protein, tau, makes the microtubules stable through a process named [[phosphorylation]] and is therefore called a [[microtubule-associated protein]].<ref name="pmid17604998">{{cite journal |author=Hernández F, Avila J |title=Tauopathies |journal=Cellular Molecular Life Sciences |volume=64 |issue=17 |pages=2219–2233 |year=2007 |pmid=17604998 |doi=10.1007/s00018-007-7220-x}}</ref> In AD, tau is changed chemically, becoming [[Hyperphosphorylation|hyperphosphorylated]].
| |
|
| |
| In the tau hypothesis, hyperphosphorylated tau begins to pair with other threads of tau and they become tangled up together inside [[nerve cell]] bodies in masses known as [[neurofibrillary tangles]].<ref name="pmid1669718">{{cite journal |author=Goedert M, Spillantini MG, Crowther RA |title=Tau proteins and neurofibrillary degeneration |journal=Brain Pathology |volume=1 |issue=4 |pages=279–286 |year=1991 |pmid=1669718 | doi=10.1111/j.1750-3639.1991.tb00671.x }}</ref> When this happens, the microtubules disintegrate, collapsing the neuron's transport system. This may result first in malfunctions in communication between [[neurons]] and later in the death of the cells.<ref name="pmid17127334">{{cite journal |author=Chun W, Johnson GV |title=The role of tau phosphorylation and cleavage in neuronal cell death |journal=Frontiers of Bioscience |volume=12 |pages=733–756 |year=2007 |pmid=17127334}}</ref>
| |
| Both [[amyloid plaques]] and [[neurofibrillary tangle]]s are clearly visible by [[microscopy]] in AD brains.<ref name="pmid15184601">{{cite journal |author=Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J |title=The importance of neuritic plaques and tangles to the development and evolution of AD |journal=Neurology |volume=62 |issue=11 |pages=1984–1989 |year=2004 |pmid=15184601 |doi=}}</ref> Plaques are dense, mostly [[insoluble]] deposits of amyloid-beta [[protein]] and [[cell]]ular material outside and around neurons. Tangles are insoluble twisted fibers that build up inside the nerve cell. Though many older people develop some plaques and tangles, the brains of AD patients have them to a much greater extent and in different brain locations.<ref name="pmid8038565">{{cite journal |author=Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH |title=Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital |journal=Cerebral Cortex |volume=4 |issue=2 |pages=138–150 |year=1994 |pmid=8038565 |doi =10.1093/cercor/4.2.138 }}</ref>
| |
|
| |
| ===Alternative hypotheses===
| |
|
| |
| Recent research supports the previously obscure theory that [[Herpes_simplex#Alzheimer.27s_disease|Herpes simplex]] virus type 1 plays a role as a possible cause of AD in people carrying the susceptible versions of the [[Apolipoprotein E|apoE]] gene.<ref name=Itzhaki2008>{{cite journal |author=Wozniak MA, Mee AP, Itzhaki RF |title=Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques |journal=J Pathol. |volume=217 |issue=1 |pages=131–8 |year=2009 |month=January |pmid=18973185 |doi=10.1002/path.2449 |url=http://www3.interscience.wiley.com/journal/121411445/abstract}}</ref>
| |
| As [[HSV-1]] is not a new virus, consideration of additional factors will be needed to explain the increase
| |
| [http://www.cdc.gov/nchs/pressroom/07newsreleases/lifeexpectancy.htm|increase] in the age-adjusted incidence of AD. Various [[inflammatory processes]] and inflammatory [[cytokines]] may also have a role in the pathology of Alzheimer's disease. However, these are general markers of tissue damage in any disease, and may also be either secondary causes of tissue damage in AD, or else bystander "marker" effects.<ref>{{cite journal |author=Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK |title=New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. |journal=Ann N Y Acad Sci.|volume=1035 |issue=Dec |pages=290–315 |year=2004 |pmid=15681814 |doi=10.1196/annals.1332.018 }}</ref> Other [[cholinergic]] effects have also been proposed—for example, the initiation of large-scale aggregation of amyloid,<ref name="pmid15236795">{{cite journal
| |
| |author=Shen ZX
| |
| |title=Brain cholinesterases: II. The molecular and cellular basis of Alzheimer's disease
| |
| |journal=Med. Hypotheses
| |
| |volume=63
| |
| |issue=2
| |
| |pages=308–21
| |
| |year=2004
| |
| |pmid=15236795
| |
| |doi=10.1016/j.mehy.2004.02.031
| |
| }}</ref> leading to generalized neuroinflammation.<ref name="pmid12934968">{{cite journal
| |
| |author=Wenk GL
| |
| |title=Neuropathologic changes in Alzheimer's disease
| |
| |journal=J Clin Psychiatry
| |
| |volume=64 Suppl 9
| |
| |pages=7–10
| |
| |year=2003
| |
| |pmid=12934968
| |
| }}</ref>
| |
|
| |
| ===Associated Conditions=== | | ===Associated Conditions=== |
|
| |
|