Fanconi anemia physical examination
|
Fanconi anemia Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Case Studies |
|
Fanconi anemia physical examination On the Web |
|
American Roentgen Ray Society Images of Fanconi anemia physical examination |
|
Risk calculators and risk factors for Fanconi anemia physical examination |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief:
Overview
CLINICAL FEATURES
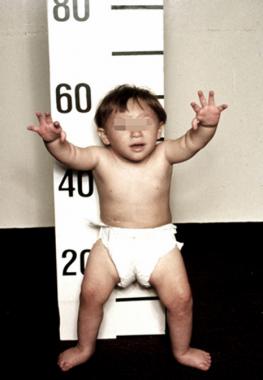
Congenital anomalies — Congenital malformations are the most common presenting features of FA. Malformations are reported in 60 to 75 percent of patients, but many in the field believe this represents an underestimate, as many patients with FA do not manifest with classical findings.
Young adults with more subtle clinical findings increasingly may be identified from genomic sequencing. Despite the high frequency of malformations, only a small percentage of patients with FA (<5 percent) are diagnosed within the first year of life based on classic congenital anomalies. Thus, while the presence of these findings provides an important clue to the diagnosis, their absence does not eliminate the possibility of FA.
In a series of 370 patients enrolled in the International FA Registry and a review of over 2000 patients reported in the literature from 1927 to 2009, the most common developmental abnormalities included the following.
● Skin findings (approximately 40 to 60 percent), including hyper- or hypopigmentation or café-au-lait spots
● Short stature (40 to 60 percent)
● Thumb or other radial ray abnormalities (50 percent)
• Thumbs absent or hypoplastic, bifid/duplicated, rudimentary, triphalangeal (35 percent)
• Radii absent or hypoplastic (7 percent)
• Hands/other such as flat thenar eminence, clinodactyly, polydactyly, missing first metacarpal, dysplastic ulnae (6 percent)
● Axial skeletal abnormalities (25 percent), especially microcephaly, triangular facies, short/webbed neck, vertebral anomalies
● Eye malformations (20 to 40 percent), including strabismus and hypo/hypertelorism
● Renal and urinary tract malformations (approximately 20 to 30 percent) including horseshoe, ectopic, dysplastic, or absent kidney; hydronephrosis; hydroureter
● Gonadal/Genital malformations
• In males, hypospadias, micropenis, undescended/absent testes, infertility (25 percent)
• In females, uterus malformation, small ovaries, hypogenitalia (<5 percent)
● Ear abnormalities (10 to 20 percent) with conductive hearing loss due to middle ear anomalies or atretic ear canal
● Congenital heart disease (approximately 5 percent) such as patent ductus arteriosus, ventricular septal defect, aortic coarctation, truncus arteriosus
● Gastrointestinal anomalies (approximately 5 percent) such as tracheoesophageal fistula, esophageal atresia, intestinal atresia, imperforate anus
● Central nervous system abnormalities (<5 percent) involving the pituitary gland (eg, small, interrupted pituitary stalk syndrome), hydrocephalus, cerebellar hypoplasia, or absent corpus callosum.
Cytopenias/bone marrow failure — Cytopenias in FA include thrombocytopenia, macrocytic anemia, or pancytopenia. Bone marrow failure eventually occurs in the majority of patients, though the time to onset can be quite variable. Progression to pancytopenia may occur rapidly after initial cytopenias are noted or may take months or years to develop, or (rarely) may not develop at all.
●Cytopenias – Cytopenias may only be mild upon presentation or may develop later in the disease course if the FA diagnosis is made based on congenital anomalies. In some cases only a single cell line will be involved (typically thrombocytopenia), particularly early in life. Mild to moderate thrombocytopenia may be misdiagnosed as immune thrombocytopenia (ITP). The anemia is typically macrocytic, and some individuals may have macrocytosis without anemia. The degree of cytopenia may be used to characterize the degree of bone marrow failure as mild, moderate, or severe (table 2). Most patients eventually develop symptomatic anemia, although contrary to the name Fanconi "anemia," symptomatic anemia is often the last severe cytopenia to develop. Severe neutropenia (absolute neutrophil count [ANC] < 500/microL) and thrombocytopenia (platelet count <30,000/microL) are often more problematic, as they can lead to potentially life-threatening infections and bleeding. Definitions of disease severity based on the bone marrow cellularity and blood counts is discussed in more detail separately. (See "Acquired aplastic anemia in children and adolescents", section on 'Definitions of severity'.)
●Bone marrow failure – In a 2003 report from the International FA Registry that included 754 patients, 601 (80 percent) had bone marrow failure at the time of enrollment; the cumulative incidence was 90 percent by age 40 [18]. In the small percentage of patients in whom bone marrow failure does not develop, specific genetic factors may protect from bone marrow aplasia. Patients with biallelic FANCD2/BRCA2mutations appear less likely to develop bone marrow failure, though the high rate and early onset of malignancies, coupled with the short lifespan in this population may contribute to this perceived effect. In addition, up to 25 percent of patients with FA may develop acquired somatic mosaicism through gene conversion events (in compound heterozygous patients), back mutation, or even compensatory deletions/insertions, which lead to correction of the chromosomal breakage sensitivity phenotype [81-84]. While many patients may have somatic mosaicism detectable only in lymphocytes, patients with mosaicism in hematopoietic stem cells (HSCs) or progenitor cells have been shown to have an ameliorated bone marrow phenotype. However, these individuals remain at risk for hematologic malignancy and other non-hematologic complications.
Findings on bone marrow examination may be indistinguishable from findings seen in other causes of bone marrow failure such as aplastic anemia or myelodysplastic syndrome (MDS). For patients diagnosed with FA in infancy due to congenital anomalies, screening bone marrow biopsies are often normocellular. By the onset of cytopenias, the marrow may reveal severe hypocellularity out of proportion to the degree of cytopenias. Erythroid dysplasia, including hyperplastic erythroblast islands and megaloblastic features, is commonly seen in many, but not all, bone marrow aspirates from patients with FA, and should not be interpreted in isolation as MDS in the absence of other dysplastic features, increased blasts, or cytogenetic changes. On the other hand, dysplasia in the myeloid series, increased myeloblasts, and with somewhat less specificity dysmegakaryopoiesis, should be considered to be concerning evidence for onset of clonal abnormalities consistent with MDS [22,85].
MDS/Leukemia — MDS and leukemia are common in patients with FA; in many cases, MDS or acute myeloid leukemia (AML) is the presenting finding. Patients with FA have been estimated to have a 6000-fold and 700-fold greater risk than the general population for developing MDS and AML, respectively [86]. By age 50, up to 40 percent of patients with FA will develop MDS and up to 15 percent will develop AML. Lymphoid malignancies including acute lymphoblastic leukemia (ALL) and Burkitt lymphoma are also seen, although they are much less common [18]. A period of bone marrow hypoplasia precedes the development of hematologic malignancies in some but not all cases [22].
Leukemia risk is even higher in patients with biallelic mutations in FANCD1/BRCA2. These individuals have a cumulative incidence of leukemia of 80 percent by age 10 [80]. Most develop AML, although some may develop T cell ALL. Patients with mutations in BRCA2 involving the IVS7 site have particularly early risk of leukemia, with most developing AML by three years of age.
Karyotypic abnormalities are common in patients with FA who develop MDS or AML, including translocations of chromosome 1p, monosomy 7, and gains of chromosome 3q [22]. In one study of 53 patients, 18 had 3q amplification, which was associated with shorter survival and increased risk for development of AML [87]. A 2012 literature review identified 46 cases of AML in patients with FA in whom cytogenetics were available and found the most common cytogenetic abnormalities to be chromosomal gains of 1q, 3q, or 13q, along with loss of chromosome 7 (or more specifically, 7q) [88]. In contrast, cytogenetic lesions common in de novo AML including t(8;21), trisomy 8, and inv(16) were not seen in any of the patients with FA.
Cytogenetic clones should be interpreted within the context of the bone marrow morphology. Some cytogenetic clones of unclear clinical significance may remain stable or become undetectable over time, whereas loss of part or all of chromosome 7 necessitates consideration of hematopoietic cell transplant (HCT) prior to leukemia progression. With any cytogenetic abnormality, close monitoring of the bone marrow and the blood counts is warranted. (See "Management and prognosis of Fanconi anemia", section on 'Hematologic neoplasms'.)
Solid tumors — A number of solid tumor types have been reported to occur at increased frequency in individuals with FA, and these appear at a much younger age than the age at which these tumors are seen in unaffected individuals. As an example, a 2003 study involving a cohort of 1300 individuals with FA estimated the median age of cancer development to be approximately 16 years, compared with 68 years in the general population [89]. In many late-onset cases of FA, malignancy is the presenting finding.
The cumulative incidence of solid tumors is increasing as individuals with FA are living longer, due to cure of bone marrow failure by HCT. In addition, HCT may increase the risk of solid tumors in some individuals with FA, likely due to a combination of factors including exposure to DNA damaging agents or radiation in the conditioning regimen and the development of graft-versus-host disease (GVHD). This trend was demonstrated in a study that compared the rates of cancer between 117 individuals with FA who underwent HCT with 145 who did not [90]. The age-specific hazard of squamous cell cancer was 4.4-fold higher in individuals who had a transplant, and the tumors occurred at a younger age (median age, 18 versus 33 years). In another series of 37 individuals with FA who underwent HCT, the 15-year incidence of head and neck cancers was 53 percent [91].
Unlike for AML, where risk in patients with FA reaches a plateau between 30 to 40 years of age, the annual risk of developing a solid tumor continues to increase significantly with age, particularly in FA patients greater than age 30 [18,86,92-94]. A 2003 study from the International FA Registry estimated the cumulative incidence of solid tumors by the age of 40 years at 28 percent [18]. This study followed 754 patients for over 20 years and identified 79 solid tumors. The most common were squamous cell cancers (SCCs) of the head, neck, esophagus, anus, and urogenital region; these accounted for 39 of the solid tumors (49 percent). There were also 18 liver tumors, accounting for 23 percent of tumors; as well as six renal tumors, five brain tumors, three breast cancers, and other tumor types including germ cell tumors and sarcomas. Similar findings have been reported in other cohorts [92,93].
Despite this high incidence of malignancy, solid tumors are rare in childhood, with the exception of those harboring biallelic FANCD1/BRCA2mutations, in whom the likelihood of at least one malignancy is greater than 97 percent by seven years of age [95]. For patients with FANCD1/BRCA2 mutations, brain tumors occur in over 50 percent by five years of age (second only to leukemia in frequency), although new onset brain tumors are rare beyond this age [86]. Wilms tumor is also common in patients with biallelic FANCD1/BRCA2 mutations, and less frequently other solid tumors of childhood are seen, including rhabdomyosarcoma and neuroblastoma [95].
The role of human papilloma virus (HPV) infection in patients with FA who develop SCC is unclear. A 2003 report from a United States cohort suggested that the high incidence of SCC of the head/neck and anal/urogenital regions in patients with FA were due to increased susceptibility to genomic instability produced by HPV, as >80 percent of these tumors were HPV-positive [96]. However, subsequent reports contradicted these findings, demonstrating HPV DNA in only two of 21 and one of nine patients in the two reports [97,98]. Many of the tumors in the European cohort demonstrated p53 mutations [98]. Thus, whether patients with FA have increased indication to receive vaccination to HPV compared to the general public remains unknown. We give the HPV vaccine to all patients with FA since uncertainty remains regarding this issue. (See "Virology of human papillomavirus infections and the link to cancer" and "Epidemiology and risk factors for head and neck cancer" and "Management and prognosis of Fanconi anemia", section on 'Solid tumors'.)
Endocrine manifestations — Individuals with FA may have a range of endocrine disorders. In many cases, endocrine abnormalities result from anatomical disruption of the hypothalamic-pituitary axis during development, including common abnormalities such as pituitary stalk interruption syndrome and septo-optic dysplasia [99]. In other cases, specific organ dysfunction, either intrinsic to the disease or as a consequence of HCT-associated therapies (eg, conditioning regimen, therapy for GVHD) leads to endocrine abnormalities.
●Short stature is seen in the majority of patients, but some patients have normal or even above-average height regardless of genotype [100]. In many cases, short stature is driven by growth hormone deficiency.
●Primary hypothyroidism is seen in over 60 percent of patients with FA, usually due to central hypothalamic or intrinsic thyroid dysfunction rather than autoimmunity.
●Adrenal dysfunction occurs in a subset of patients due to low ACTH secretion, although these patients will generally have a normal response to exogenous ACTH stimulation [100].
●Altered glucose metabolism, including diabetes mellitus and impaired glucose tolerance, occurs in nearly 50 percent of patients with FA due to dysfunction of pancreatic islet cells [101].
●Patients with FA are also at increased risk for dyslipidemia and other aspects of metabolic syndrome.
●Infertility and delayed or abnormal progression of puberty are also very frequent in FA. In males, infertility may result from gonadal dysfunction and/or developmental defects in genital tract formation. In females fertility is possible; however, premature ovarian failure occurs in over 75 percent of patients [102].
Physical Examination
- Congenital malformations are the most common presenting features of FA.
OR
- Physical examination of patients with [disease name] is usually remarkable for:[finding 1], [finding 2], and [finding 3].
- The presence of [finding(s)] on physical examination is diagnostic of [disease name].
- The presence of [finding(s)] on physical examination is highly suggestive of [disease name].
Appearance of the Patient
- Patients with [disease name] usually appear [general appearance].
Vital Signs
- High-grade / low-grade fever
- Hypothermia / hyperthermia may be present
- Tachycardia with regular pulse or (ir)regularly irregular pulse
- Bradycardia with regular pulse or (ir)regularly irregular pulse
- Tachypnea / bradypnea
- Kussmal respirations may be present in _____ (advanced disease state)
- Weak/bounding pulse / pulsus alternans / paradoxical pulse / asymmetric pulse
- High/low blood pressure with normal pulse pressure / wide pulse pressure / narrow pulse pressure
Skin
- Skin examination of patients with [disease name] is usually normal.
OR
-
Description (Adapted from Dermatology Atlas)
-
Description (Adapted from Dermatology Atlas)

{kind=link}
HEENT
- HEENT examination of patients with [disease name] is usually normal.
OR
- Abnormalities of the head/hair may include ___
- Evidence of trauma
- Icteric sclera
- Nystagmus
- Extra-ocular movements may be abnormal
- Pupils non-reactive to light / non-reactive to accommodation / non-reactive to neither light nor accommodation
- Ophthalmoscopic exam may be abnormal with findings of ___
- Hearing acuity may be reduced
- Weber test may be abnormal (Note: A positive Weber test is considered a normal finding / A negative Weber test is considered an abnormal finding. To avoid confusion, you may write "abnormal Weber test".)
- Rinne test may be positive (Note: A positive Rinne test is considered a normal finding / A negative Rinne test is considered an abnormal finding. To avoid confusion, you may write "abnormal Rinne test".)
- Exudate from the ear canal
- Tenderness upon palpation of the ear pinnae/tragus (anterior to ear canal)
- Inflamed nares / congested nares
- Purulent exudate from the nares
- Facial tenderness
- Erythematous throat with/without tonsillar swelling, exudates, and/or petechiae
Neck
- Neck examination of patients with [disease name] is usually normal.
OR
- Jugular venous distension
- Carotid bruits may be auscultated unilaterally/bilaterally using the bell/diaphragm of the otoscope
- Lymphadenopathy (describe location, size, tenderness, mobility, and symmetry)
- Thyromegaly / thyroid nodules
- Hepatojugular reflux
Lungs
- Pulmonary examination of patients with [disease name] is usually normal.
OR
- Asymmetric chest expansion / Decreased chest expansion
- Lungs are hypo/hyperresonant
- Fine/coarse crackles upon auscultation of the lung bases/apices unilaterally/bilaterally
- Rhonchi
- Vesicular breath sounds / Distant breath sounds
- Expiratory/inspiratory wheezing with normal / delayed expiratory phase
- Wheezing may be present
- Egophony present/absent
- Bronchophony present/absent
- Normal/reduced tactile fremitus
Heart
- Cardiovascular examination of patients with [disease name] is usually normal.
OR
- Chest tenderness upon palpation
- PMI within 2 cm of the sternum (PMI) / Displaced point of maximal impulse (PMI) suggestive of ____
- Heave / thrill
- Friction rub
- S1
- S2
- S3
- S4
- Gallops
- A high/low grade early/late systolic murmur / diastolic murmur best heard at the base/apex/(specific valve region) may be heard using the bell/diaphgram of the otoscope
Abdomen
Abdominal examination of patients with [disease name] is usually normal.
OR
- Abdominal distention
- Abdominal tenderness in the right/left upper/lower abdominal quadrant
- Rebound tenderness (positive Blumberg sign)
- A palpable abdominal mass in the right/left upper/lower abdominal quadrant
- Guarding may be present
- Hepatomegaly / splenomegaly / hepatosplenomegaly
- Additional findings, such as obturator test, psoas test, McBurney point test, Murphy test
Back
- Back examination of patients with [disease name] is usually normal.
OR
- Point tenderness over __ vertebrae (e.g. L3-L4)
- Sacral edema
- Costovertebral angle tenderness bilaterally/unilaterally
- Buffalo hump
Genitourinary
- Gonads may display the following abnormalities:
- Males - Hypogenitalia, undescended testes, hypospadias, abnormal or absent testis, atrophic testes, azoospermia, phimosis, abnormal urethra, micropenis, delayed development
- Females - Hypogenitalia; bicornuate uterus; aplasia of uterus and vagina; atresia of uterus, vagina, or ovary/ovaries
Neuromuscular
- Neck - Sprengel abnormality, short, low hairline, webbed
- Spine - Spina bifida (thoracic, lumbar, cervical, occult sacral), scoliosis, abnormal ribs, sacrococcygeal sinus, Klippel-Feil syndrome, vertebral anomalies, extra vertebrae
Extremities
Upper limb abnormalities can include the following features:
- Thumbs - Absent or hypoplastic, supernumerary, bifid, rudimentary, short, low set, attached by a thread, triphalangeal, tubular, stiff, hyperextensible
- Radii - Absent or hypoplastic (only with abnormal thumbs [ie, terminal defects]), absent or weak pulse
- Hands - Clinodactyly, hypoplastic thenar eminence, 6 fingers, absent first metacarpal, enlarged abnormal fingers, short fingers
- Ulnae - Dysplastic
- Lower limb Abnormalities can include the following features:
- Feet - Toe syndactyly, abnormal toes, flat feet, short toes, clubfoot, 6 toes
- Legs - Congenital hip dislocation, Perthes disease, coxa vara, abnormal femur, thigh osteoma, abnormal legsL