Epoprostenol
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]; Associate Editor(s)-in-Chief: Gerald Chi
Disclaimer
WikiDoc MAKES NO GUARANTEE OF VALIDITY. WikiDoc is not a professional health care provider, nor is it a suitable replacement for a licensed healthcare provider. WikiDoc is intended to be an educational tool, not a tool for any form of healthcare delivery. The educational content on WikiDoc drug pages is based upon the FDA package insert, National Library of Medicine content and practice guidelines / consensus statements. WikiDoc does not promote the administration of any medication or device that is not consistent with its labeling. Please read our full disclaimer here.
Overview
Epoprostenol is a prostaglandin that is FDA approved for the treatment of pulmonary arterial hypertension. Common adverse reactions include bradyarrhythmia, chest pain, hypotension, tachycardia, flushing, abdominal pain, diarrhea, loss of appetite, nausea, vomiting, dizziness, and headache.
Adult Indications and Dosage
FDA-Labeled Indications and Dosage (Adult)
Pulmonary Arterial Hypertension
- Important Note: Epoprostenol sodium for injection must be reconstituted only with STERILE DILUENT for epoprostenol sodium for injection. Do not dilute reconstituted solutions of epoprostenol for injection or administer with other parenteral solutions or medications.
- Dosing Information
- Administer continuous chronic infusion of epoprostenol sodium for injection through a central venous catheter. Temporary peripheral intravenous infusion may be used until central access is established. Initiate chronic infusion of epoprostenol sodium for injection at 2 ng/kg/min and increase in increments of 2 ng/kg/min every 15 minutes or longer until dose-limiting pharmacologic effects are elicited or until a tolerance limit to the drug is established or further increases in the infusion rate are not clinically warranted. If dose-limiting pharmacologic effects occur, then decrease the infusion rate until epoprostenol sodium for injection is tolerated. In clinical trials, the most common dose-limiting adverse events were nausea, vomiting, hypotension, sepsis, headache, abdominal pain, or respiratory disorder (most treatment-limiting adverse events were not serious). If the initial infusion rate of 2 ng/kg/min is not tolerated, identify a lower dose that is tolerated by the patient.
- In the controlled 12-week trial in PAH/SSD, for example, the dose increased from a mean starting dose of 2.2 ng/kg/min. During the first 7 days of treatment, the dose was increased daily to a mean dose of 4.1 ng/kg/min on day 7 of treatment. At the end of week 12, the mean dose was 11.2 ng/kg/min. The mean incremental increase was 2 to 3 ng/kg/min every 3 weeks.
- Dosage Adjustments
- Base changes in the chronic infusion rate on persistence, recurrence, or worsening of the patient's symptoms of pulmonary hypertension and the occurrence of adverse events due to excessive doses of epoprostenol sodium for injection. In general, expect increases in dose from the initial chronic dose.
- Consider increments in dose if symptoms of PAH persist or recur. Increase the infusion by 1- to 2-ng/kg/min increments at intervals sufficient to allow assessment of clinical response; these intervals should be at least 15 minutes. In clinical trials, incremental increases in dose occurred at intervals of 24 to 48 hours or longer. Following establishment of a new chronic infusion rate, observe the patient, and monitor standing and supine blood pressure and heart rate for several hours to ensure that the new dose is tolerated.
- During chronic infusion, the occurrence of dose-limiting pharmacological events may necessitate a decrease in infusion rate, but the adverse event may occasionally resolve without dosage adjustment. Make dosage decreases gradually in 2-ng/kg/min decrements every 15 minutes or longer until the dose-limiting effects resolve. Avoid abrupt withdrawal of epoprostenol sodium for injection or sudden large reductions in infusion rates. Except in life-threatening situations (e.g., unconsciousness, collapse, etc.), adjust infusion rates of epoprostenol sodium for injection only under the direction of a physician.
- In patients receiving lung transplants, doses of epoprostenol sodium for injection were tapered after the initiation of cardiopulmonary bypass.
Off-Label Use and Dosage (Adult)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Epoprostenol in adult patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Epoprostenol in adult patients.
Pediatric Indications and Dosage
FDA-Labeled Indications and Dosage (Pediatric)
- Safety and effectiveness in pediatric patients have not been established.
Off-Label Use and Dosage (Pediatric)
Guideline-Supported Use
There is limited information regarding Off-Label Guideline-Supported Use of Epoprostenol in pediatric patients.
Non–Guideline-Supported Use
There is limited information regarding Off-Label Non–Guideline-Supported Use of Epoprostenol in pediatric patients.
Contraindications
- A large study evaluating the effect of epoprostenol on survival in NYHA Class III and IV patients with congestive heart failure due to severe left ventricular systolic dysfunction was terminated after an interim analysis of 471 patients revealed a higher mortality in patients receiving epoprostenol plus conventional therapy than in those receiving conventional therapy alone. The chronic use of epoprostenol in patients with congestive heart failure due to severe left ventricular systolic dysfunction is therefore contraindicated.
- Some patients with pulmonary hypertension have developed pulmonary edema during dose initiation, which may be associated with pulmonary veno-occlusive disease. Epoprostenol should not be used chronically in patients who develop pulmonary edema during dose initiation.
- Epoprostenol is also contraindicated in patients with known hypersensitivity to the drug or to structurally related compounds.
Warnings
- Epoprostenol sodium for injection must be reconstituted only as directed using STERILE DILUENT for epoprostenol sodium for injection. Epoprostenol sodium for injection must not be reconstituted or mixed with any other parenteral medications or solutions prior to or during administration.
Abrupt Withdrawal
- Abrupt withdrawal (including interruptions in drug delivery) or sudden large reductions in dosage of epoprostenol may result in symptoms associated with rebound pulmonary hypertension, including dyspnea, dizziness, and asthenia. In clinical trials, one Class III patient's death was judged attributable to the interruption of epoprostenol. Avoid abrupt withdrawal.
Precautions
- Epoprostenol should be used only by clinicians experienced in the diagnosis and treatment of pulmonary hypertension. Carefully establish the diagnosis of idiopathic or heritable PAH or PAH/CTD.
- Epoprostenol is a potent pulmonary and systemic vasodilator. Initiate epoprostenol in a setting with adequate personnel and equipment for physiologic monitoring and emergency care. Dose initiation has been performed during right heart catheterization and without cardiac catheterization. During dose initiation, asymptomatic increases in pulmonary artery pressure coincident with increases in cardiac output occurred rarely. In such cases, consider dose reduction, but such an increase does not imply that chronic treatment is contraindicated.
- Epoprostenol is a potent inhibitor of platelet aggregation. Therefore, expect an increased risk for hemorrhagic complications, particularly for patients with other risk factors for bleeding.
- During chronic use, deliver epoprostenol continuously on an ambulatory basis through a permanent indwelling central venous catheter. Unless contraindicated, administer anticoagulant therapy to patients receiving epoprostenol to reduce the risk of pulmonary thromboembolism or systemic embolism through a patent foramen ovale. To reduce the risk of infection, use aseptic technique in the reconstitution and administration of epoprostenol and in routine catheter care. Because epoprostenol is metabolized rapidly, even brief interruptions in the delivery of epoprostenol may result in symptoms associated with rebound pulmonary hypertension including dyspnea, dizziness, and asthenia. Intravenous therapy with epoprostenol will likely be needed for prolonged periods, possibly years, so consider the patient's ability to accept and care for a permanent intravenous catheter and infusion pump.
- Based on clinical trials, the acute hemodynamic response to epoprostenol did not correlate well with improvement in exercise tolerance or survival during chronic use of epoprostenol. Adjust dosage of epoprostenol during chronic use at the first sign of recurrence or worsening of symptoms attributable to pulmonary hypertension or the occurrence of adverse events associated with epoprostenol. Following dosage adjustments, monitor standing and supine blood pressure and heart rate closely for several hours.
Adverse Reactions
Clinical Trials Experience
- During clinical trials, adverse events were classified as follows: (1) adverse events during dose initiation and escalation, (2) adverse events during chronic dosing, and (3) adverse events associated with the drug delivery system.
Adverse Events During Dose Initiation and Escalation
- During early clinical trials, epoprostenol was increased in 2-ng/kg/min increments until the patients developed symptomatic intolerance. The most common adverse events and the adverse events that limited further increases in dose were generally related to vasodilation, the major pharmacologic effect of epoprostenol. The most common dose-limiting adverse events (occurring in ≥1% of patients) were nausea, vomiting, headache, hypotension, and flushing, but also include chest pain, anxiety, dizziness, bradycardia, dyspnea, abdominal pain, musculoskeletal pain, and tachycardia. Table 3 lists the adverse events reported during dose initiation and escalation in decreasing order of frequency.
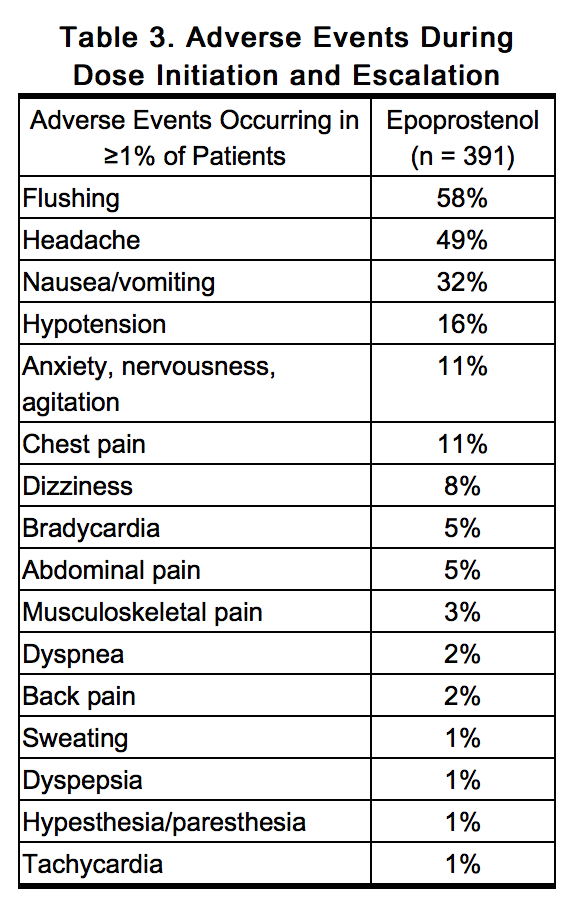
This image is provided by the National Library of Medicine.

Adverse Events During Chronic Administration
- Interpretation of adverse events is complicated by the clinical features of PAH, which are similar to some of the pharmacologic effects of epoprostenol (e.g., dizziness, syncope). Adverse events which may be related to the underlying disease include dyspnea, fatigue, chest pain, edema, hypoxia, right ventricular failure, and pallor. Several adverse events, on the other hand, can clearly be attributed to epoprostenol. These include hypotension, bradycardia, tachycardia, pulmonary edema, bleeding at various sites, thrombocytopenia, headache, abdominal pain, pain (unspecified), sweating, rash, arthralgia, jaw pain, flushing, diarrhea, nausea and vomiting, flu-like symptoms, anxiety/nervousness, and agitation. In addition, chest pain, fatigue, and pallor have been reported during epoprostenol therapy, and a role for the drug in these events cannot be excluded.
Adverse Events During Chronic Administration for Idiopathic or Heritable PAH
- In an effort to separate the adverse effects of the drug from the adverse effects of the underlying disease, Table 4 lists adverse events that occurred at a rate at least 10% greater on epoprostenol in controlled trials.
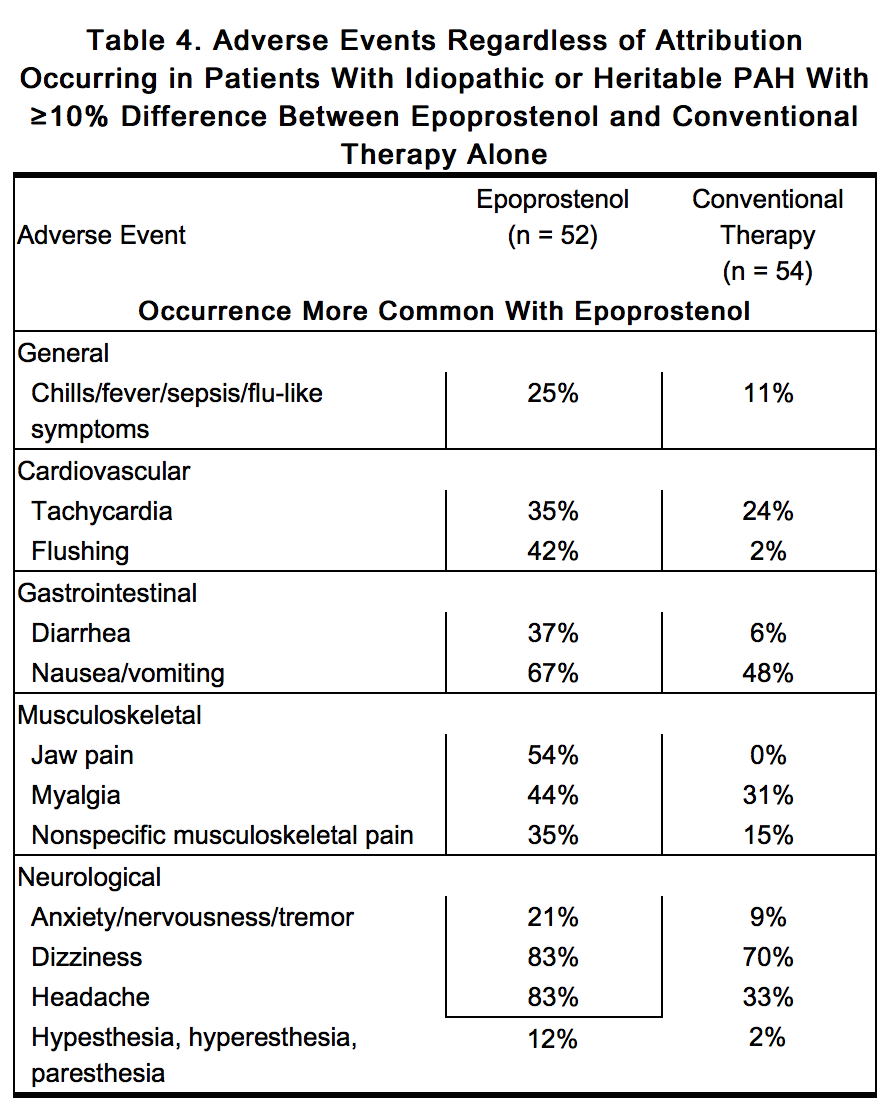
This image is provided by the National Library of Medicine.

- Thrombocytopenia has been reported during uncontrolled clinical trials in patients receiving epoprostenol.
Adverse Events During Chronic Administration for PAH/SSD
- In an effort to separate the adverse effects of the drug from the adverse effects of the underlying disease, Table 5 lists adverse events that occurred at a rate at least 10% greater on epoprostenol in the controlled trial.

This image is provided by the National Library of Medicine.

- Although the relationship to epoprostenol administration has not been established, pulmonary embolism has been reported in several patients taking epoprostenol and there have been reports of hepatic failure.
Adverse Events Attributable to the Drug Delivery System
- Chronic infusions of epoprostenol are delivered using a small, portable infusion pump through an indwelling central venous catheter. During controlled PAH trials of up to 12 weeks’ duration, the local infection rate was about 18%, and the rate for pain was about 11%. During long-term follow-up, sepsis was reported at a rate of 0.3 infections/patient per year in patients treated with epoprostenol. This rate was higher than reported in patients using chronic indwelling central venous catheters to administer parenteral nutrition, but lower than reported in oncology patients using these catheters. Malfunctions in the delivery system resulting in an inadvertent bolus of or a reduction in epoprostenol were associated with symptoms related to excess or insufficient epoprostenol, respectively.
Observed During Clinical Practice
- In addition to adverse reactions reported from clinical trials, the following events have been identified during post-approval use of epoprostenol. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These events have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to epoprostenol.
- Blood and Lymphatic: Anemia, hypersplenism, pancytopenia, splenomegaly.
- Endocrine and Metabolic: Hyperthyroidism.
Postmarketing Experience
There is limited information regarding Postmarketing Experience of Epoprostenol in the drug label.
Drug Interactions
- Additional reductions in blood pressure may occur when epoprostenol is administered with diuretics, antihypertensive agents, or other vasodilators. When other antiplatelet agents or anticoagulants are used concomitantly, there is the potential for epoprostenol to increase the risk of bleeding. However, patients receiving infusions of epoprostenol in clinical trials were maintained on anticoagulants without evidence of increased bleeding. In clinical trials, epoprostenol was used with digoxin, diuretics, anticoagulants, oral vasodilators, and supplemental oxygen.
- In a pharmacokinetic substudy in patients with congestive heart failure receiving furosemide or digoxin in whom therapy with epoprostenol was initiated, apparent oral clearance values for furosemide (n = 23) and digoxin (n = 30) were decreased by 13% and 15%, respectively, on the second day of therapy and had returned to baseline values by day 87. The change in furosemide clearance value is not likely to be clinically significant. However, patients on digoxin may show elevations of digoxin concentrations after initiation of therapy with epoprostenol, which may be clinically significant in patients prone to digoxin toxicity.
Use in Specific Populations
Pregnancy
- Pregnancy Category B
- Reproductive studies have been performed in pregnant rats and rabbits at doses up to 100 mcg/kg/day (600 mcg/m2/day in rats, 2.5 times the recommended human dose, and 1,180 mcg/m2/day in rabbits, 4.8 times the recommended human dose based on body surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to epoprostenol. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
- Australian Drug Evaluation Committee (ADEC) Pregnancy Category
There is no Australian Drug Evaluation Committee (ADEC) guidance on usage of Epoprostenol in women who are pregnant.
Labor and Delivery
- The use of epoprostenol during labor, vaginal delivery, or cesarean section has not been adequately studied in humans.
Nursing Mothers
- It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when epoprostenol is administered to a nursing woman.
Pediatric Use
- Safety and effectiveness in pediatric patients have not been established.
Geriatic Use
- Clinical studies of epoprostenol in pulmonary hypertension did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
Gender
There is no FDA guidance on the use of Epoprostenol with respect to specific gender populations.
Race
There is no FDA guidance on the use of Epoprostenol with respect to specific racial populations.
Renal Impairment
There is no FDA guidance on the use of Epoprostenol in patients with renal impairment.
Hepatic Impairment
There is no FDA guidance on the use of Epoprostenol in patients with hepatic impairment.
Females of Reproductive Potential and Males
There is no FDA guidance on the use of Epoprostenol in women of reproductive potentials and males.
Immunocompromised Patients
There is no FDA guidance one the use of Epoprostenol in patients who are immunocompromised.
Administration and Monitoring
Administration
- Intravenous
- Epoprostenol sodium for injection is administered by continuous intravenous infusion via a central venous catheter using an ambulatory infusion pump. During initiation of treatment, epoprostenol sodium for injection may be administered peripherally.
- The ambulatory infusion pump used to administer epoprostenol sodium for injection should: (1) be small and lightweight, (2) be able to adjust infusion rates in 2-ng/kg/min increments, (3) have occlusion, end-of-infusion, and low-battery alarms, (4) be accurate to ±6% of the programmed rate, and (5) be positive pressure-driven (continuous or pulsatile) with intervals between pulses not exceeding 3 minutes at infusion rates used to deliver epoprostenol sodium for injection. The reservoir should be made of polyvinyl chloride, polypropylene, or glass. The infusion pump used in the most recent clinical trials was the CADD-1 HFX 5100 (SIMS Deltec). A 60-inch microbore non-DEHP extension set with proximal antisyphon valve, low priming volume (0.9 mL), and in-line 0.22 micron filter was used during clinical trials.
- To avoid interruptions in drug delivery, the patient should have access to a backup infusion pump and intravenous infusion sets. Consider a multi-lumen catheter if other intravenous therapies are routinely administered.
- To facilitate extended use at ambient temperatures exceeding 20° to 25°C (68° to 77°F), a cold pouch with frozen gel packs was used in clinical trials. The cold pouches and gel packs used in clinical trials were obtained from Palco Labs, Palo Alto, California. Any cold pouch used must be capable of maintaining the temperature of reconstituted epoprostenol sodium for injection between 2° and 8°C for 12 hours.
Reconstitution
- Epoprostenol sodium for injection is stable only when reconstituted with STERILE DILUENT for epoprostenol sodium for injection. Epoprostenol sodium for injection must not be reconstituted or mixed with any other parenteral medications or solutions prior to or during administration.
- Select a concentration for the solution of epoprostenol sodium for injection that is compatible with the infusion pump being used with respect to minimum and maximum flow rates, reservoir capacity, and the infusion pump criteria listed above. * When administered chronically, prepare epoprostenol sodium for injection in a drug delivery reservoir appropriate for the infusion pump with a total reservoir volume of at least 100 mL, using 2 vials of STERILE DILUENT for epoprostenol sodium for injection for use during a 24-hour period. Table 6 gives directions for preparing several different concentrations of epoprostenol sodium for injection.
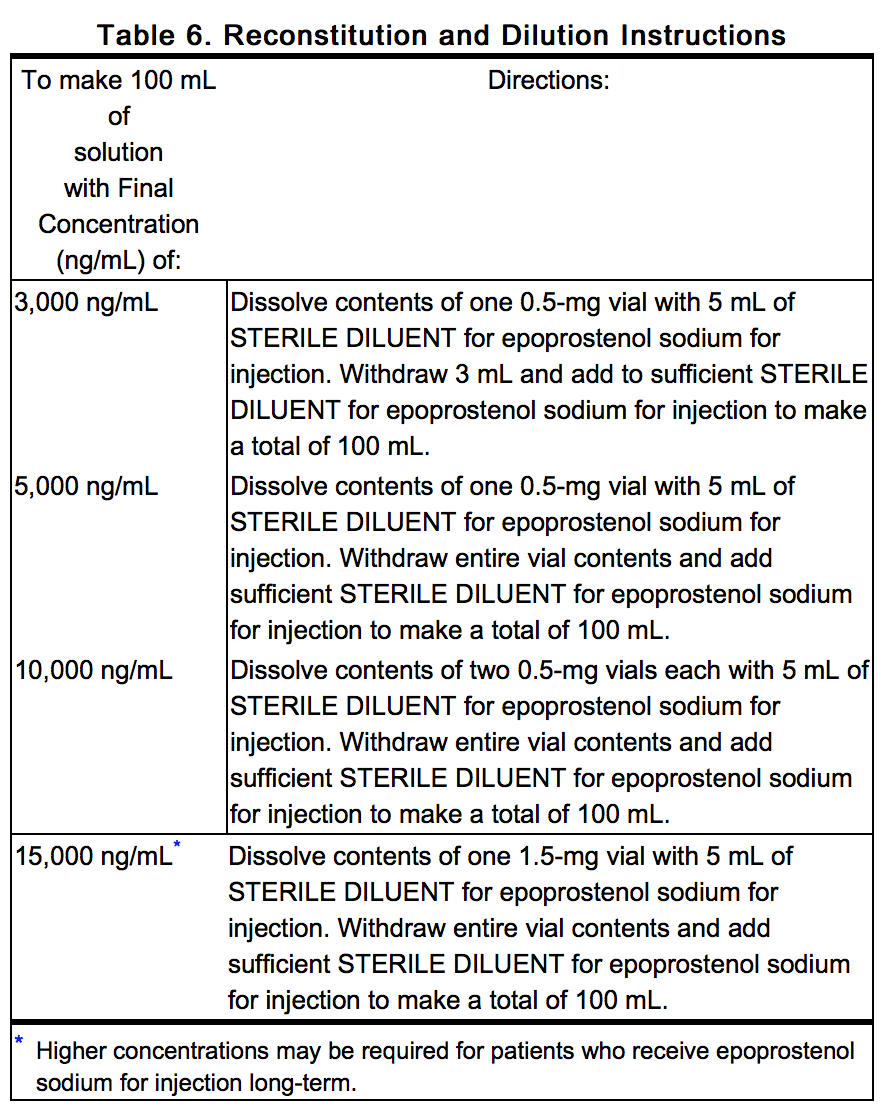
This image is provided by the National Library of Medicine.

- Generally, 3,000 ng/mL and 10,000 ng/mL are satisfactory concentrations to deliver between 2 to 16 ng/kg/min in adults. Infusion rates may be calculated using the following formula:

This image is provided by the National Library of Medicine.

- Tables 7 through 10 provide infusion delivery rates for doses up to 16 ng/kg/min based upon patient weight, drug delivery rate, and concentration of the solution of epoprostenol sodium for injection to be used. These tables may be used to select the most appropriate concentration of epoprostenol sodium for injection that will result in an infusion rate between the minimum and maximum flow rates of the infusion pump and that will allow the desired duration of infusion from a given reservoir volume. Higher infusion rates, and therefore, more concentrated solutions may be necessary with long-term administration of epoprostenol sodium for injection.
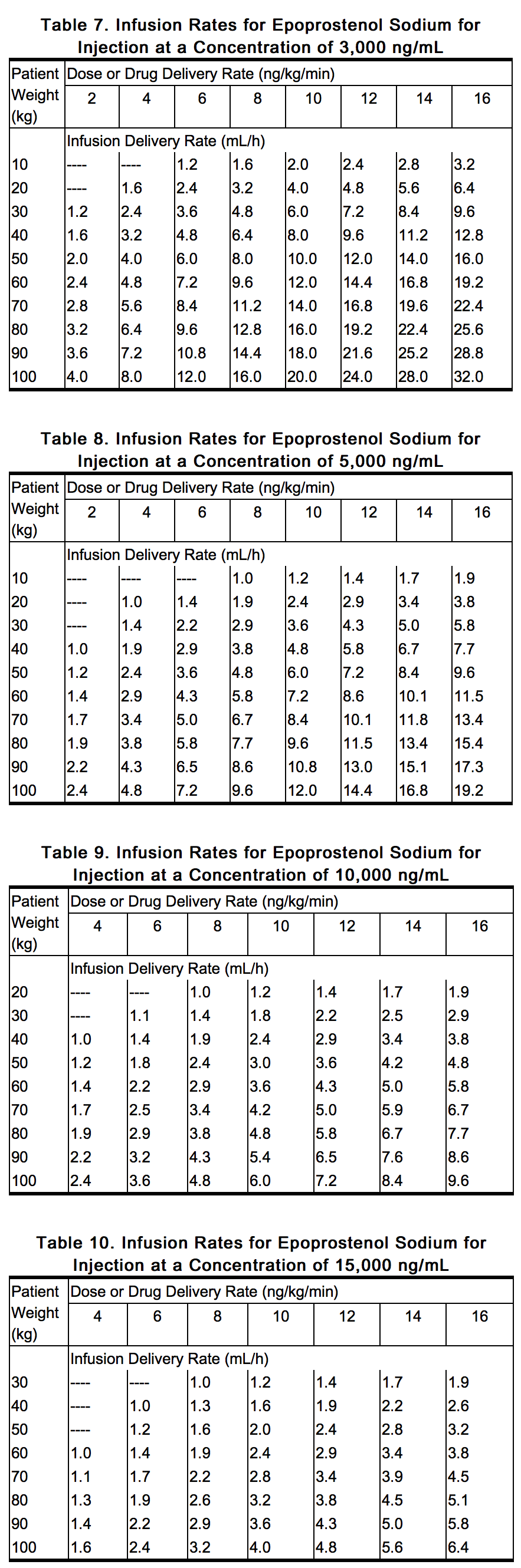
This image is provided by the National Library of Medicine.

- Use at Room Temperature
- Prior to use at room temperature, 15° to 25°C (59° to 77°F), reconstituted solutions of epoprostenol sodium for injection may be stored refrigerated at 2° to 8°C (36° to 46°F) for no longer than 40 hours. When administered at room temperature, reconstituted solutions may be used for no longer than 8 hours. This 48-hour period allows the patient to reconstitute a 2-day supply (200 mL) of epoprostenol sodium for injection. Each 100 mL daily supply may be divided into 3 equal portions. Two of the portions are stored refrigerated at 2° to 8°C (36° to 46°F) until they are used.
- Use with a Cold Pouch
- Prior to infusion with the use of a cold pouch, solutions may be stored refrigerated at 2° to 8°C (36° to 46°F) for up to 24 hours. When a cold pouch is employed during the infusion, reconstituted solutions of epoprostenol sodium for injection may be used for no longer than 24 hours. Change gel packs every 12 hours. Reconstituted solutions may be kept at 2° to 8°C (36° to 46°F), either in refrigerated storage or in a cold pouch or a combination of the two, for no more than 48 hours.
- Inspect parenteral drug products for particulate matter and discoloration prior to administration whenever solution and container permit. If either occurs, do not administer.
Monitoring
There is limited information regarding Monitoring of Epoprostenol in the drug label.
IV Compatibility
There is limited information regarding IV Compatibility of Epoprostenol in the drug label.
Overdosage
Acute Overdose
- Signs and symptoms of excessive doses of epoprostenol during clinical trials are the expected dose-limiting pharmacologic effects of epoprostenol, including flushing, headache, hypotension, tachycardia, nausea, vomiting, and diarrhea. Treatment will ordinarily require dose reduction of epoprostenol.
- One patient with PAH/CTD accidentally received 50 mL of an unspecified concentration of epoprostenol. The patient vomited and became unconscious with an initially unrecordable blood pressure. Epoprostenol was discontinued and the patient regained consciousness within seconds. In clinical practice, fatal occurrences of hypoxemia, hypotension, and respiratory arrest have been reported following overdosage of epoprostenol.
- Single intravenous doses of epoprostenol at 10 and 50 mg/kg (2,703 and 27,027 times the recommended acute phase human dose based on body surface area) were lethal to mice and rats, respectively. Symptoms of acute toxicity were hypoactivity, ataxia, loss of righting reflex, deep slow breathing, and hypothermia.
Chronic Overdose
There is limited information regarding Chronic Overdose of Epoprostenol in the drug label.
Pharmacology

| |

| |
Epoprostenol
| |
| Systematic (IUPAC) name | |
| (Z)-5-[(4R,5R)-5-hydroxy-4-((S,E)-3-hydroxyoct-1-enyl)hexahydro-2H-cyclopenta[b]furan-2-ylidene]pentanoic acid | |
| Identifiers | |
| CAS number | |
| ATC code | B01 |
| PubChem | |
| DrugBank | |
| Chemical data | |
| Formula | Template:OrganicBox atomTemplate:OrganicBox atomTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBox atomTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBoxTemplate:OrganicBox |
| Mol. mass | 352.465 g/mol |
| SMILES | & |
| Pharmacokinetic data | |
| Bioavailability | ? |
| Metabolism | ? |
| Half life | ? |
| Excretion | ? |
| Therapeutic considerations | |
| Pregnancy cat. |
? |
| Legal status | |
| Routes | ? |
Mechanism of Action
- Epoprostenol has 2 major pharmacological actions: (1) direct vasodilation of pulmonary and systemic arterial vascular beds, and (2) inhibition of platelet aggregation. In animals, the vasodilatory effects reduce right- and left-ventricular afterload and increase cardiac output and stroke volume. The effect of epoprostenol on heart rate in animals varies with dose. At low doses, there is vagally mediated bradycardia, but at higher doses, epoprostenol causes reflex tachycardia in response to direct vasodilation and hypotension. No major effects on cardiac conduction have been observed. Additional pharmacologic effects of epoprostenol in animals include bronchodilation, inhibition of gastric acid secretion, and decreased gastric emptying.
Structure
- Epoprostenol sodium for injection is a sterile sodium salt formulated for intravenous (IV) administration. Each vial of epoprostenol sodium for injection contains epoprostenol sodium equivalent to either 0.5 mg (500,000 ng) or 1.5 mg (1,500,000 ng) epoprostenol, 3.76 mg glycine, 2.93 mg sodium chloride, and 50 mg mannitol. Sodium hydroxide may have been added to adjust pH.
- Epoprostenol (PGI2, PGX, prostacyclin), a metabolite of arachidonic acid, is a naturally occurring prostaglandin with potent vasodilatory activity and inhibitory activity of platelet aggregation.
- Epoprostenol is (5Z,9α,11α,13E,15S)-6,9-epoxy-11,15-dihydroxyprosta-5,13-dien-1-oic acid.
- Epoprostenol sodium has a molecular weight of 374.45 and a molecular formula of C20H31NaO5. The structural formula is:
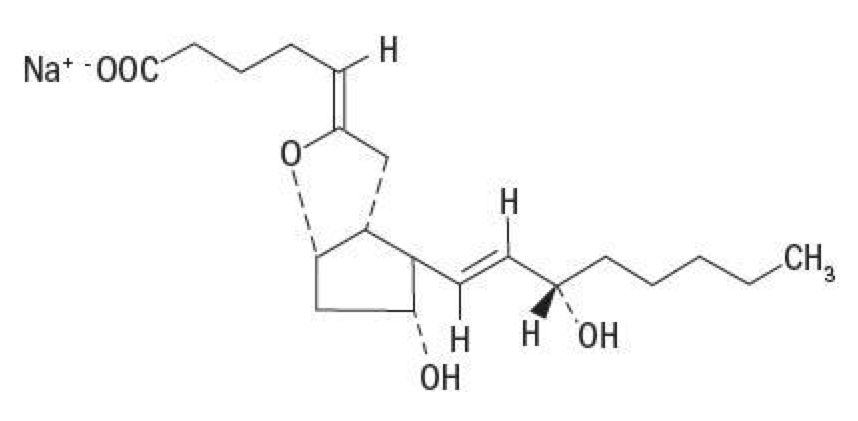
This image is provided by the National Library of Medicine.

- Epoprostenol sodium for injection is a white to off-white powder that must be reconstituted with STERILE DILUENT for epoprostenol sodium for injection. STERILE DILUENT for epoprostenol sodium for injection is supplied in glass vials containing 50 mL of 94 mg glycine, 73.3 mg sodium chloride, sodium hydroxide (added to adjust pH), and Water for Injection, USP.
- The reconstituted solution of epoprostenol sodium for injection has a pH of 11.0 to 11.8 and is increasingly unstable at a lower pH.
Pharmacodynamics
There is limited information regarding Pharmacodynamics of Epoprostenol in the drug label.
Pharmacokinetics
- Epoprostenol is rapidly hydrolyzed at neutral pH in blood and is also subject to enzymatic degradation. Animal studies using tritium-labeled epoprostenol have indicated a high clearance (93 mL/kg/min), small volume of distribution (357 mL/kg), and a short half-life (2.7 minutes). During infusions in animals, steady-state plasma concentrations of tritium-labeled epoprostenol were reached within 15 minutes and were proportional to infusion rates.
- No available chemical assay is sufficiently sensitive and specific to assess the in vivo human pharmacokinetics of epoprostenol. The in vitro half-life of epoprostenol in human blood at 37°C and pH 7.4 is approximately 6 minutes; therefore, the in vivo half-life of epoprostenol in humans is expected to be no greater than 6 minutes. The in vitro pharmacologic half-life of epoprostenol in human plasma, based on inhibition of platelet aggregation, was similar for males (n = 954) and females (n = 1,024).
- Tritium-labeled epoprostenol has been administered to humans in order to identify the metabolic products of epoprostenol. Epoprostenol is metabolized to 2 primary metabolites: 6-keto-PGF1α (formed by spontaneous degradation) and 6,15-diketo-13,14-dihydro-PGF1α (enzymatically formed), both of which have pharmacological activity orders of magnitude less than epoprostenol in animal test systems. The recovery of radioactivity in urine and feces over a 1-week period was 82% and 4% of the administered dose, respectively. Fourteen additional minor metabolites have been isolated from urine, indicating that epoprostenol is extensively metabolized in humans.
Nonclinical Toxicology
- Carcinogenesis, Mutagenesis, Impairment of Fertility
- Long-term studies in animals have not been performed to evaluate carcinogenic potential. A micronucleus test in rats revealed no evidence of mutagenicity. The Ames test and DNA elution tests were also negative, although the instability of epoprostenol makes the significance of these tests uncertain. Fertility was not impaired in rats given epoprostenol by subcutaneous injection at doses up to 100 mcg/kg/day (600 mcg/m2/day, 2.5 times the recommended human dose [4.6 ng/kg/min or 245.1 mcg/m2/day, IV] based on body surface area).
Clinical Studies
- Acute Hemodynamic Effects
- Acute intravenous infusions of epoprostenol for up to 15 minutes in patients with idiopathic or heritable PAH or PAH associated with scleroderma spectrum of diseases (PAH/SSD) produce dose-related increases in cardiac index (CI) and stroke volume (SV) and dose-related decreases in pulmonary vascular resistance (PVR), total pulmonary resistance (TPR), and mean systemic arterial pressure (SAPm). The effects of epoprostenol on mean pulmonary artery pressure (PAPm) were variable and minor.
Chronic Infusion in Idiopathic or Heritable PAH
- Hemodynamic Effects
- Chronic continuous infusions of epoprostenol in patients with idiopathic or heritable PAH were studied in 2 prospective, open, randomized trials of 8 and 12 weeks’ duration comparing epoprostenol plus conventional therapy to conventional therapy alone. Dosage of epoprostenol was determined as described in DOSAGE AND ADMINISTRATION and averaged 9.2 ng/kg/min at study’s end. Conventional therapy varied among patients and included some or all of the following: anticoagulants in essentially all patients; oral vasodilators, diuretics, and digoxin in one half to two thirds of patients; and supplemental oxygen in about half the patients. Except for 2 New York Heart Association (NYHA) functional Class II patients, all patients were either functional Class III or Class IV. As results were similar in the 2 studies, the pooled results are described.
- Chronic hemodynamic effects were generally similar to acute effects. Increases in CI, SV, and arterial oxygen saturation and decreases in PAPm, mean right atrial pressure (RAPm), TPR, and systemic vascular resistance (SVR) were observed in patients who received epoprostenol chronically compared to those who did not. Table 1 illustrates the treatment-related hemodynamic changes in these patients after 8 or 12 weeks of treatment.
- These hemodynamic improvements appeared to persist when epoprostenol was administered for at least 36 months in an open, nonrandomized study.

This image is provided by the National Library of Medicine.

- Clinical Effects
- Statistically significant improvement was observed in exercise capacity, as measured by the 6-minute walk test in patients receiving continuous intravenous epoprostenol plus conventional therapy (N = 52) for 8 or 12 weeks compared to those receiving conventional therapy alone (N = 54). Improvements were apparent as early as the first week of therapy. Increases in exercise capacity were accompanied by statistically significant improvement in dyspnea and fatigue, as measured by the Chronic Heart Failure Questionnaire and the Dyspnea Fatigue Index.
- Survival was improved in NYHA functional Class III and Class IV patients with idiopathic or heritable PAH treated with epoprostenol for 12 weeks in a multicenter, open, randomized, parallel study. At the end of the treatment period, 8 of 40 (20%) patients receiving conventional therapy alone died, whereas none of the 41 patients receiving epoprostenol died (p = 0.003).
Chronic Infusion in PAH/Scleroderma Spectrum of Diseases (SSD)
- Hemodynamic Effects
- Chronic continuous infusions of epoprostenol in patients with PAH/SSD were studied in a prospective, open, randomized trial of 12 weeks’ duration comparing epoprostenol plus conventional therapy (N = 56) to conventional therapy alone (N = 55). Except for 5 NYHA functional Class II patients, all patients were either functional Class III or Class IV. Dosage of epoprostenol was determined as described in DOSAGE AND ADMINISTRATION and averaged 11.2 ng/kg/min at study’s end. Conventional therapy varied among patients and included some or all of the following: anticoagulants in essentially all patients, supplemental oxygen and diuretics in two thirds of the patients, oral vasodilators in 40% of the patients, and digoxin in a third of the patients. A statistically significant increase in CI, and statistically significant decreases in PAPm, RAPm, PVR, and SAPm after 12 weeks of treatment were observed in patients who received epoprostenol chronically compared to those who did not. Table 2 illustrates the treatment-related hemodynamic changes in these patients after 12 weeks of treatment.
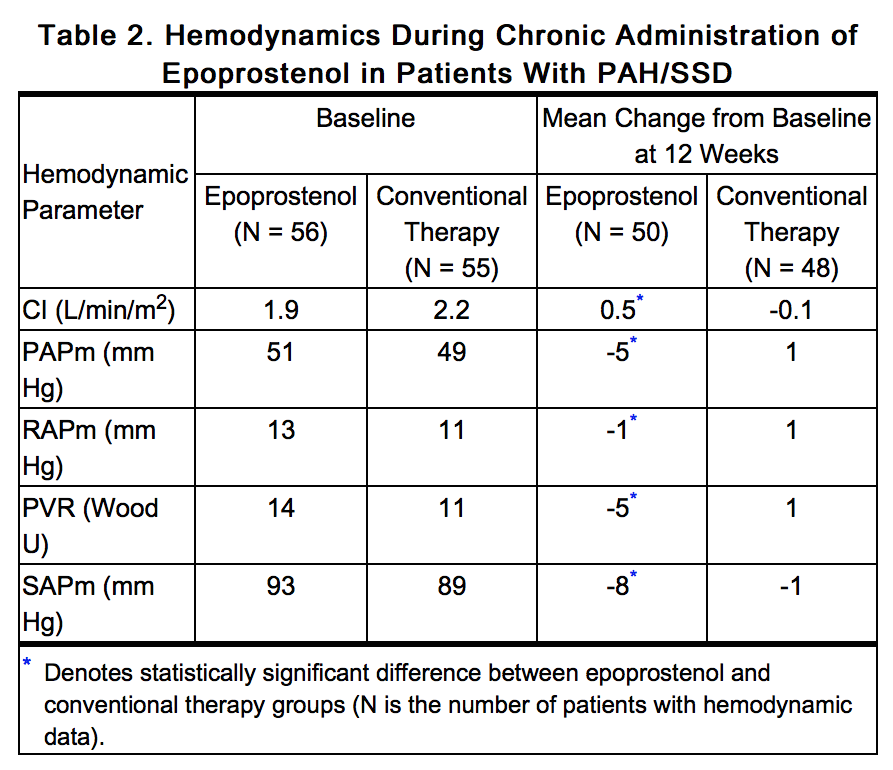
This image is provided by the National Library of Medicine.

- Clinical Effects
- Statistically significant improvement was observed in exercise capacity, as measured by the 6-minute walk, in patients receiving continuous intravenous epoprostenol plus conventional therapy for 12 weeks compared to those receiving conventional therapy alone. Improvements were apparent in some patients at the end of the first week of therapy. Increases in exercise capacity were accompanied by statistically significant improvements in dyspnea and fatigue, as measured by the Borg Dyspnea Index and Dyspnea Fatigue Index. At week 12, NYHA functional class improved in 21 of 51 (41%) patients treated with epoprostenol compared to none of the 48 patients treated with conventional therapy alone. However, more patients in both treatment groups (28/51 [55%] with epoprostenol and 35/48 [73%] with conventional therapy alone) showed no change in functional class, and 2/51 (4%) with epoprostenol and 13/48 (27%) with conventional therapy alone worsened. Of the patients randomized, NYHA functional class data at 12 weeks were not available for 5 patients treated with epoprostenol and 7 patients treated with conventional therapy alone.
- No statistical difference in survival over 12 weeks was observed in PAH/SSD patients treated with epoprostenol as compared to those receiving conventional therapy alone. At the end of the treatment period, 4 of 56 (7%) patients receiving epoprostenol died, whereas 5 of 55 (9%) patients receiving conventional therapy alone died.
- No controlled clinical trials with epoprostenol have been performed in patients with pulmonary hypertension associated with other diseases.
How Supplied
- Epoprostenol sodium for injection is supplied as a sterile freeze-dried powder in 10 mL flint glass vials with gray butyl rubber closures, individually packaged in a carton.

This image is provided by the National Library of Medicine.

- Store the vials of epoprostenol sodium for injection at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F). Protect from light. Retain in carton until time of use. Discard unused portion.
- The STERILE DILUENT for Epoprostenol Sodium for Injection is supplied in flint glass vials containing 50 mL diluent with butyl rubber closures.

This image is provided by the National Library of Medicine.

- Storage and Stability
- Unopened vials of epoprostenol sodium for injection are stable until the date indicated on the package when stored at 20° to 25°C (68° to 77°F) and protected from light in the carton. Unopened vials of STERILE DILUENT for epoprostenol sodium for injection are stable until the date indicated on the package when stored at 20° to 25°C (68° to 77°F).
- Prior to use, reconstituted solutions of epoprostenol sodium for injection must be protected from light and must be refrigerated at 2° to 8°C (36° to 46°F) if not used immediately. Do not freeze reconstituted solutions of epoprostenol sodium for injection. Discard any reconstituted solution that has been frozen. Discard any reconstituted solution if it has been refrigerated for more than 48 hours.
- During use, a single reservoir of reconstituted solution of epoprostenol sodium for injection can be administered at room temperature for a total duration of 8 hours, or it can be used with a cold pouch and administered up to 24 hours with the use of 2 frozen 6 oz gel packs in a cold pouch. When stored or in use, insulate reconstituted epoprostenol sodium for injection from temperatures greater than 25°C (77°F) and less than 0°C (32°F), and do not expose to direct sunlight.
Storage
There is limited information regarding Epoprostenol Storage in the drug label.
Images
Drug Images
{{#ask: Page Name::Epoprostenol |?Pill Name |?Drug Name |?Pill Ingred |?Pill Imprint |?Pill Dosage |?Pill Color |?Pill Shape |?Pill Size (mm) |?Pill Scoring |?NDC |?Drug Author |format=template |template=DrugPageImages |mainlabel=- |sort=Pill Name }}
Package and Label Display Panel
{{#ask: Label Page::Epoprostenol |?Label Name |format=template |template=DrugLabelImages |mainlabel=- |sort=Label Page }}
Patient Counseling Information
- Patients receiving epoprostenol should receive the following information. Epoprostenol sodium for injection must be reconstituted only with STERILE DILUENT for epoprostenol sodium for injection. Epoprostenol is infused continuously through a permanent indwelling central venous catheter via a small, portable infusion pump. Thus, therapy with epoprostenol requires commitment by the patient to drug reconstitution, drug administration, and care of the permanent central venous catheter. Patients must adhere to sterile technique in preparing the drug and in the care of the catheter, and even brief interruptions in the delivery of epoprostenol may result in rapid symptomatic deterioration. A patient’s decision to receive epoprostenol should be based upon the understanding that there is a high likelihood that therapy with epoprostenol will be needed for prolonged periods, possibly years. The patient's ability to accept and care for a permanent intravenous catheter and infusion pump should also be carefully considered.
Precautions with Alcohol
- Alcohol-Epoprostenol interaction has not been established. Talk to your doctor about the effects of taking alcohol with this medication.
Brand Names
- Veletri®
- Flolan®[1]
Look-Alike Drug Names
- N/A[2]
Drug Shortage Status
Price
References
The contents of this FDA label are provided by the National Library of Medicine.
- ↑ "EPOPROSTENOL SODIUM injection, powder, for solution".
- ↑ "http://www.ismp.org". External link in
|title=(help)
{{#subobject:
|Page Name=Epoprostenol
|Pill Name=No image.jpg
|Drug Name=
|Pill Ingred=|+sep=;
|Pill Imprint=
|Pill Dosage={{{dosageValue}}} {{{dosageUnit}}}
|Pill Color=|+sep=;
|Pill Shape=
|Pill Size (mm)=
|Pill Scoring=
|Pill Image=
|Drug Author=
|NDC=
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol12.png
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol13.png
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol14.png
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol15.png
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol16.png
}}
{{#subobject:
|Label Page=Epoprostenol |Label Name=Epoprostenol17.png
}}