Diffuse axonal injury
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
Diffuse axonal injury (DAI) is one of the most common and devastating types of brain injury (Iwata et al., 2004), occurring in almost half of all cases of severe head trauma (Park and Hyun, 2004). It is a type of diffuse brain injury, meaning that damage occurs over a more widespread area than in focal brain injury. DAI, which refers to extensive lesions in white matter tracts, is one of the major causes of unconsciousness and persistent vegetative state after head trauma (Wasserman, 2004).
Though diffuse axonal injury seldom leads to death, the outcome is frequently coma with over 90% of patients with severe DAI never regaining consciousness (Wasserman, 2004). Those who do wake up often remain significantly impaired (Vinas and Pilistis, 2004).
Nowadays, other authors state that DAI can occur in every degree of severity from (very) mild or moderate to (very) severe (Vik, 2006, Smith, 2000).
Mechanism
Unlike brain trauma that occurs due to direct impact and deformation of the brain, DAI is the result of traumatic shearing forces that occur when the head is rapidly accelerated or decelerated, as may occur in auto accidents, falls, and assaults (reviewed in Wolf et al., 2001). It usually results from twisting or rotational forces (angular momentum), rather than forward and back impacts linear momentum (Sanders and McKenna, 2001; Wasserman, 2004; Shepherd, 2004). Car accidents are the most frequent causes of DAI, with sports accidents and child abuse also common causes (Smith and Greenwald, 2003).
The major cause of damage in DAI is the tearing of axons, the neural processes that allow one neuron to communicate with another. Tracts of axons, which appear white due to myelination, are referred to as white matter. Acceleration causes shearing injury, which refers to damage inflicted as tissue slides over other tissue. When the brain is accelerated, parts of differing densities and distances from the axis of rotation slide over one another, stretching axons that traverse junctions between areas of different density, especially gray-white matter junctions (Wasserman, 2004). Two thirds of DAI lesions occur in areas where grey and white matter meet (Wasserman, 2004).
Characteristics
Patients typically have several focal white matter lesions of variable size (1-15 mm) in a characteristic distribution. Areas most vulnerable to injury are the frontal and temporal lobes (Boone and de Montfort, 2002). Other common locations for DAI include the white matter in the cerebral cortex, the corpus callosum, the superior cerebral peduncles, basal ganglia, thalamus, and deep hemispheric nuclei (Smith and Greenwald, 2003; Stock and Singer, 2004). These areas may be more easily damaged because of the difference in density between them and the rest of the brain (Stock and Singer, 2004).
Histological characteristics
DAI is characterized by axonal separation, in which the axon is torn at the site of stretch and the part distal to the tear degrades. While it was once thought that the main cause of axonal separation was tearing due to mechanical forces during the trauma, it is now understood that secondary biochemical cascades, which occur in response to the primary injury and take place hours to days after the initial injury, are largely responsible for the damage to axons (Wolf et al., 2001; Arundine et al., 2004).
Though the processes involved in secondary brain injury are still poorly understood, it is now accepted that stretching of axons during injury causes physical disruption to and proteolytic degradation of the cytoskeleton (Iwata et al., 2004). It also opens sodium channels in the axolemma, which causes voltage-gated calcium channels to open and Ca2+ to flow into the cell (Iwata et al., 2004). The intracellular presence of Ca2+ unleashes several different pathways, including activating phospholipases and proteolytic enzymes, damaging mitochondria and the cytoskeleton, and activating secondary messengers, which can lead to separation of the axon and death of the cell (Wolf et al., 2001).
Cytoskeleton disruption
Axons are normally elastic, but when rapidly stretched they become brittle, and the axonal cytoskeleton can be broken. It is thought that integrins connected to the extracellular matrix outside the cell and to the cytoskeleton within it can transmit forces from the matrix to the cytoskeleton and cause the latter to tear when the axon is stretched (Ochs et al., 1996).
Misalignment of cytoskeletal elements after stretch injury can lead to tearing of the axon and death of the neuron. Axonal transport continues up to the point of the break in the cytoskeleton, but no further, leading to a buildup of transport products and local swelling at that point (Oztas, 2003). When it becomes large enough, swelling can tear the axon at the site of the break in the cytoskeleton, causing it to draw back toward the cell body and form a bulb (Smith and Meaney, 2000). This bulb is called a retraction ball, the hallmark of diffuse axonal injury (Wasserman, 2004).
When the axon is transected, Wallerian degeneration, in which the part of the axon distal to the break degrades, takes place within one to two days after injury (Lopachin and Lehning, 1997). The axolemma disintegrates (Lopachin and Lehning, 1997), myelin breaks down and begins to detach from cells in an anterograde direction (Cowie and Stanton, 2005), and nearby cells begin phagocytic activity, engulfing debris (Hughes et al., 2002).
Calcium influx
While sometimes only the cytoskeleton is disturbed, frequently disruption of the axolemma occurs as well, causing the influx of Ca2+ into the cell and unleashing a variety of degrading processes (Povlishock and Pettus, 1996; Lopachin and Lehning, 1997). An increase in Ca2+ and Na+ levels and a drop in K+ levels is found within the axon directly after injury (Lopachin and Lehning, 1997; Wolf et al., 2001). Possible routes of Ca2+ entry include sodium channels, pores torn in the membrane during stretch, and failure of ATP-dependent transporters due to mechanical blockage or lack of energy (Wolf et al., 2001). High levels of intracellular Ca2+, the major cause of post-injury cell damage (Zhou et al., 2001), destroy mitochondria (Smith and Meaney, 2000), contribute to the generation of reactive oxygen species (Arundine et al., 2004) and trigger phospholipases and proteolytic enzymes that damage Na+ channels and degrade or alter the cytoskeleton and the axoplasm (Lopachin and Lehning, 1997; Castillo and Babson, 1998). Excess Ca2+ can also lead to damage to the blood brain barrier and swelling of the brain (Zhou et al., 2001).
One of the proteins activated by the presence of calcium in the cell is calpain, a Ca2+-dependent non-lysosomal protease (Castillo and Babson, 1998). About 15 minutes to half an hour after the onset of injury, a process called calpain-mediated spectrin proteolysis, or CMSP, begins to occur (Büki et al., 2000). Calpain breaks down a molecule called spectrin, which holds the membrane onto the cytoskeleton, causing the formation of blebs and the breakdown of the cytoskeleton and the membrane, and ultimately the death of the cell (Castillo and Babson, 1998; Büki et al., 2000). Other molecules that can be degraded by calpains are microtubule subunits, microtubule-associated proteins, and neurofilaments (Castillo and Babson, 1998).
Generally occurring one to six hours into the process of post-stretch injury, the presence of calcium in the cell initiates the caspase cascade, a process in cell injury that usually leads to apoptosis, or "cell suicide" (Büki et al., 2000).
Mitochondria, dendrites, and parts of the cytoskeleton damaged in the injury have a limited ability to heal and regenerate, a process which occurs over 2 or more weeks (Corbo and Tripathi, 2004; Wasserman, 2004). After the injury, astrocytes can shrink, causing parts of the brain to atrophy (Wasserman, 2004).
Diagnosis and treatment
DAI is difficult to detect since it does not show up well on CT scans or with other macroscopic imaging techniques, though it shows up microscopically (Wasserman, 2004). Axonal damage in DAI is largely a result of secondary biochemical cascades, and has a delayed onset, so a person with DAI who initially appears well may deteriorate later. Thus injury is frequently more severe than is realized, and medical professionals must suspect DAI in any patients whose CT scans appear normal but who have symptoms like unconsciousness (Wasserman, 2004).
MRI is more sensitive: 30% of head injured patients with normal head CT scans have signs of DAI on MRI (Corbo and Tripathi, 2004). But MRI may also miss DAI, because it identifies the injury using signs of edema, which may not be present (Corbo and Tripathi, 2004).
DAI is classified in grades based on severity of the injury. In Grade I, widespread axonal damage is present but no focal abnormalities are seen. In Grade II, damage found in Grade I is present in addition to focal abnormalities, especially in the corpus callosum. Grade III damage encompasses both Grades I and II in addition to rostral brain stem injury and often tears in the tissue (Bigler, 2000).
DAI currently lacks a specific treatment beyond what is done for any type of head injury, including stabilizing the patient and trying to limit increases in intracranial pressure (ICP).
Future treatments
The fact that a single stimulus, the stretching of axons, leads to harm to the cell by the activation of multiple degradative pathways introduces possibilities for treating head injury shortly after the event and preventing a large part of the damage from occurring.
Since chemicals used to block proteolytic enzymes and Na+ and Ca2+ channels have prevented damage due to the influx of Ca2+, new drugs could be invented to mitigate damage using these chemicals. In addition, since damage occurs as a cascade, with one element of the process activating the next, a single drug that inhibits one part of a cascade could thereby block all of the products downstream from it (Arundine et al., 2004).
One must also wonder if vasoactive intestinal peptide (VIP) might one day play an important role in preventing the widespread Wallerian degeneration commonly seen in DAI. Animal and human studies have shown VIP increases the breakdown of glycogen by astrocytes, diminishes the inflammatory response, and may even promote the differentiation of oligodendrocyte precursors by agonizing prolactin secretion.
-

-
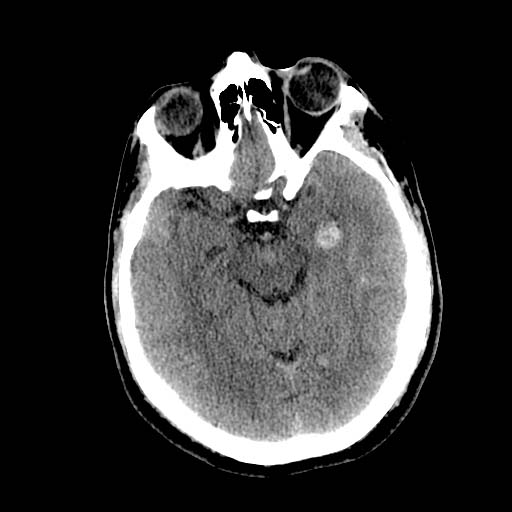
-

-
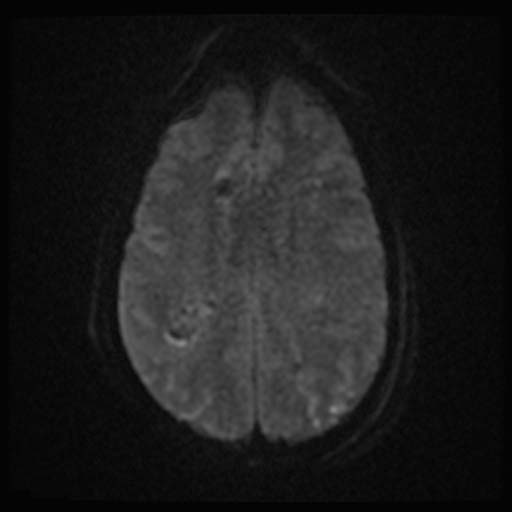
DWI
-
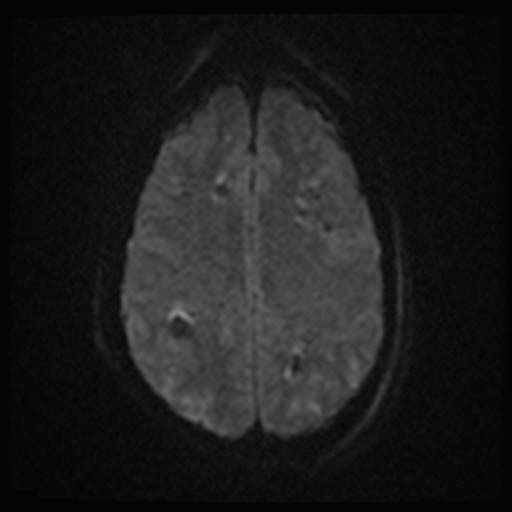
DWI Image:
-
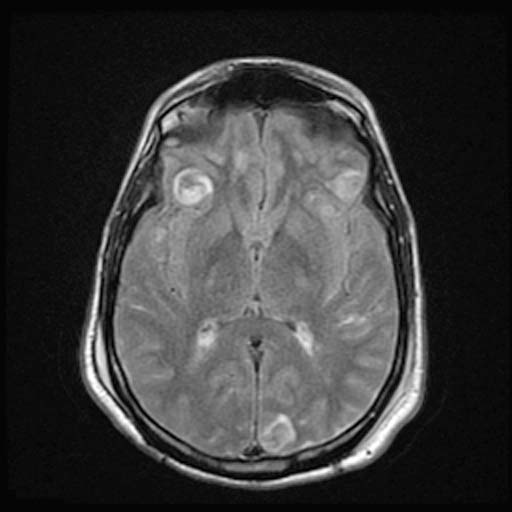
FLAIR
-
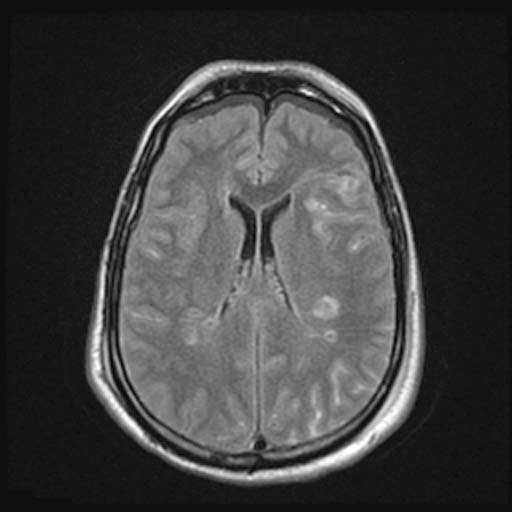
FLAIRImage:
-
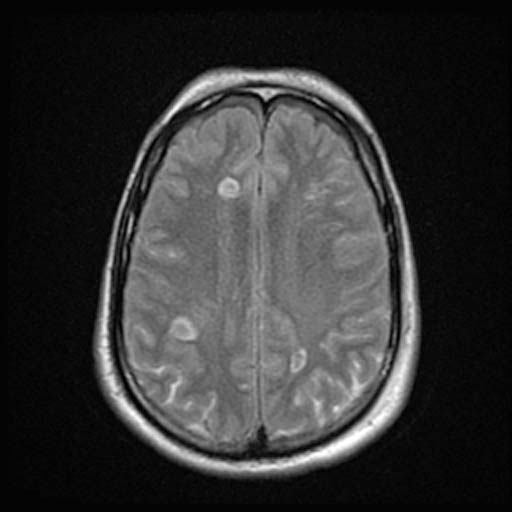
FLAIRImage:
-
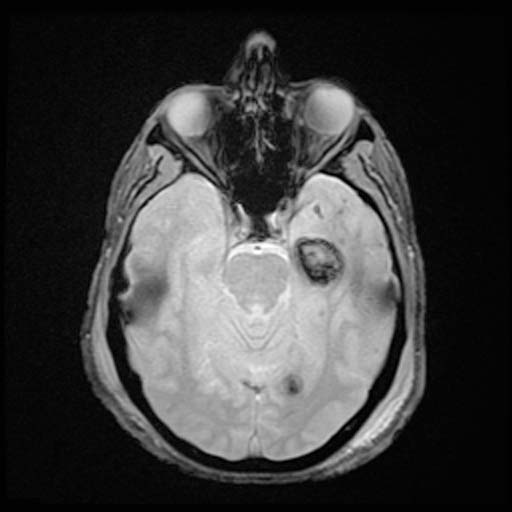
GRE
-
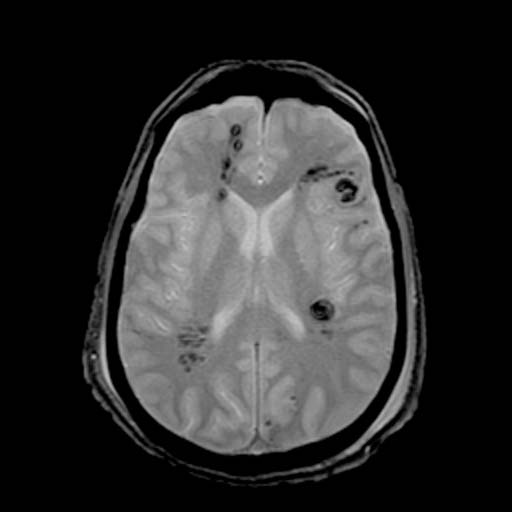
GRE
-
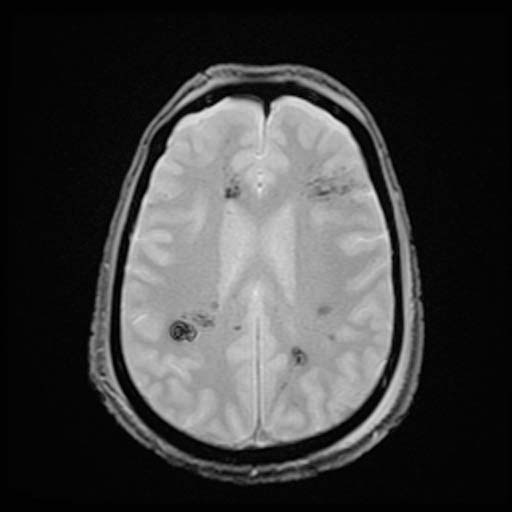
GRE











Related Chapters
External links
- TBI Resource Guide Traumatic Brain Injury Resource Guide
- Fact sheets on brain injury, its effects, and strategies for survivors and their families
- Emedicine.com: Diffuse Axonal Injury (DAI). An article on Diffuse Axonal Injury by Jeffrey R Wasserman, giving a generalized overview of Diffuse Axonal Injury (DAI), its pathophysiology and other relevant information.
- Diffuse Axonal Injury MRI and CT Images
References
- Arundine M., Aarts M., Lau A., and Tymianski M. (2004). Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. Journal of Neuroscience 24(37): 8106-8123.
- Bigler, ED. 2000. The Lesion(s) in Traumatic Brain Injury: Implications for Clinical Neuropsychology. Accessed through web archive. Retrieved March 16, 2007.
- Boon R. and de Montfor G.J. 2002. Brain Injury. Learning Discoveries Psychological Services. Learningdiscoveries.org.
- Büki A, Okonkwo D. O., Wang K. K. W., and Povlishock J. T. (2000). Cytochrome c Release and Caspase Activation in Traumatic Axonal Injury. Journal of Neuroscience. 20(8): 2825-2834.
- Castillo M. R. and Babson J. R. (1998). Ca2+-dependent mechanisms of cell injury in cultured cortical neurons. Neuroscience. 86(4): 1133-1144.
- Corbo J, Tripathi P. 2004. Delayed Presentation of Diffuse Axonal Injury: A Case Report. Trauma. 44(1).
- Cowie R. J. and Stanton G. B. (2005). Axoplasmic Transport and Neuronal Responses to Injury. Howard University College of Medicine.
- Hughes P. M., Wells G. M. A., Perry V. H., Brown M. C., and Miller K. M. (2002). [Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems]. Neuroscience. 113(2): 273-287.
- Iwata A., Stys P. K., Wolf J. A., Chen X. H., Taylor, A. G., Meaney D. F., and Smith D. H. (2004). Traumatic Axonal Injury Induces Proteolytic Cleavage of the Voltage-Gated Sodium Channels Modulated by Tetrodotoxin and Protease Inhibitors. The Journal of Neuroscience. 24(19): 4605-4613.
- LoPachin R. M. and Lehning E. J. (1997). Mechanism of Calcium Entry during Axon Injury and Degeneration. Toxicology and Applied Pharmacology. 143(2): 233-244.
- Ochs S., Pourmand R., and Jersild R. A. Jr. (1996). Origin of Beading Constrictions at the Axolemma: Presence in Unmyelinated Axons and after Beta, Beta′-Iminodipropionitrile Degradation of the Cytoskeleton. Neuroscience. 70(4): 1081-1096.
- Park C. O. and Hyun D. K. (2004). Apoptotic change in response to magnesium therapy after moderate diffuse axonal injury in rats. Yonsei Medical Journal. 45(5): 908-916.
- Povlishock J. T. and Pettus E. H. (1996). Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochirurgica. 66:81-6.
- Sanders MJ and McKenna K. 2001. Mosby’s Paramedic Textbook, 2nd revised Ed. Chapter 22, "Head and Facial Trauma." Mosby.
- Shepherd S. 2004. Head Trauma. Emedicine.com.
- Smith D. and Greenwald B. 2003. Management and Staging of Traumatic Brain Injury. Emedicine.com.
- Smith, D. H. and Meaney D. F. (2000). Axonal Damage in Traumatic Brain Injury. The Neuroscientist. 6(6): 483-495
- Stock A and Singer L. 2004. Head Trauma. Emedicine.com.
- Vik A., Kvistad, K.A., Skandsen, T. & Ingebritsen, T. (2006). Diffus aksonal skade ved hodetraume. Tiddskr. Nor. Lægeforen. 126: 2940-4.
- Vinas F.C. and Pilitsis J. 2004. Penetrating Head Trauma. Emedicine.com.
- Wasserman J. (2004). Diffuse Axonal Injury. Emedicine.com.
- Wolf J. A., Stys P. K., Lusardi T., Meaney D., and Smith, D. H. (2001). Traumatic Axonal Injury Induces Calcium Influx Modulated by Tetrodotoxin-Sensitive Sodium Channels. Journal of Neuroscience. 21(6):1923-1930
- Zhou F., Xiang Z., Feng W. X., and Zhen L. X. (2001). BBB permeability and ultrastructure in head injury with secondary insult Neuronal free Ca2+ and BBB permeability and ultrastructure in head injury with secondary insult. Journal of Clinical Neuroscience. 8(6):561-563.